
MDCG 2024-10 Rev.0
Clinical evaluation of orphan medical devices
Disclaimer: This document is an interactive version of the original MDCG document. We will keep it up-to-date.
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.
Table of Contents
1. Abbreviations and terminology
For the purposes of this guidance document, the below terms are defined as follows:
Active Implantable Medical Devices Directive, referring to Directive 90/385/EEC
MDR Article 2(24)
Clinical evaluation plan
Clinical evaluation report
MDR Article 2(53) – may be direct or indirect (see MDCG 2020-6)
MDR Article 2 (48)
MDR Article 2 (44)
MDR Article 2 (51)
MDR Article 2 (45)
MDR Article 2 (52)
MDR Article 2 (3)
General Safety and Performance Requirements, per MDR Annex I
A device that is manufactured and used only within a health institution established in the Union and that meets all condition set in Article 5(5) of the MDR or IVDR (per MDCG 2023-1)
MDR Article 2(12)
In Vitro Diagnostic Medical Devices Regulation, referring to Regulation (EU) 2017/746
Device previously CE marked under Directives 93/42/EEC (MDD) or 90/385/EEC (AIMDD) and placed on the market or put into service after the MDR’s date of application pursuant to Article 120 MDR (per MDCG 2021-25)
Medical Device Coordination Group
Medical Devices Regulation, referring to Regulation (EU) 2017/745
Medical Devices Directive, referring to Directive 93/42/EEC
Any relevant data that does not meet the MDR definition of clinical data
Refers to a device, indication, or (sub)population which does not meet the definition of “orphan device”, “orphan indication”, or “orphan (sub)population”, respectively
Device as described in section 4.1 of this document
As described in section 4.3 of this document
As described in section 4.2.1 of this document
As described in section 4.2.1 of this document
MDR Article 2(22)
Post market clinical follow-up – MDR Annex XIV, Part B, section 5
Post market surveillance – MDR Article 2(60)
MDR Article 2(23)
Devices belonging to the same generic device group (per MDR Article 2(7)). The MDR defines this as a set of devices having the same or similar intended purposes or a commonality of technology allowing them to be classified in a generic manner not reflecting specific characteristics (per MDCG 2020-6)
Group of individuals for which the medical device is intended
2.Introduction
The level of clinical evidence that is required to place medical devices on the market has been increased by the MDR, including an increased need for pre-market clinical investigations for certain higher risk devices to verify their safety and clinical performance. These increased clinical evidence requirements present a challenge for devices specifically intended for use in rare diseases/conditions, or in specific indications for rare cohorts of patients with an otherwise non-rare disease/condition.
By their nature, these ‘orphan devices’ are only intended for use in a small number of individuals each year. Many rare diseases have very few diagnostic or therapeutic options and the orphan device can be particularly crucial to fulfil an otherwise unmet medical need. In the absence of specific guidance for these devices, different understandings can emerge among manufacturers, notified bodies, and regulators on the clinical evidence requirements for these devices for the purposes of MDR certification.
In many cases, orphan devices are intended for use solely or predominantly in minors and paediatric populations, and/or in emergency situations. Proactively generating clinical data within an appropriate time in small patient populations is particularly challenging, as is the case for vulnerable populations in light of the ethical and regulatory requirements to appropriately protect these populations (1), as well as greater practical challenges of performing clinical studies in certain cohorts such as infants and children.
The increased, and at times unpredictable, financial costs associated with compliance with MDR requirements, including MDR certification, can make it prohibitive for manufacturers to place orphan devices on the EU market, as the low volumes of sales may not offset the financial costs.
Manufacturers also develop devices that have both ‘orphan’ and ‘non-orphan’ indications, used in separate and distinct population cohorts. For these devices, challenges may emerge in demonstrating sufficient pre-market clinical evidence for their orphan indication, similar to those challenges experienced for orphan devices. In such circumstances, where duly justified and without prejudice to the clinical evidence requirements for the device’s non-orphan indications, the principles outlined in this guidance are applicable to these devices for the purposes of supporting their orphan indications only.
Given their unique challenges, and in the absence of specific provisions in the MDR, the application of MDR requirements to orphan devices should be balanced and proportionate in light of Article 35 of the Charter of Fundamental Rights (health care) (2), so that the pre-market clinical evidence requirements are sufficiently met without unduly hindering or delaying patient access to these important devices. As is described in section 5 of this document, there are circumstances where it is acceptable to place an orphan device on the market with limitations in pre-market clinical data. In such circumstances, it is important that there is an appropriate level of transparency for health care professionals, patients, and members of the public, so that they are aware of the orphan status of the device, any relevant limitations in clinical data, and any relevant conditions or provisions of certification that have been applied.
In their August 2022 position paper (3), the MDCG acknowledged the unique challenges facing orphan devices and the need to develop specific guidance for these devices. To that end, the MDCG convened a dedicated orphan device task force, which led the development of this guidance document in collaboration with stakeholder representatives including notified bodies, industry, academic societies, and healthcare professionals.
To frame this guidance and to highlight the devices for which this guidance is intended, criteria have been developed that a device needs to meet to qualify for orphan device status. These criteria reflect the quantitative and qualitative characteristics of an orphan device, referring respectively to the relevant epidemiology, and to the insufficiency of alternatives and expected clinical benefit.
This document is divided into two parts, and includes guidance on the following:
PART A – Clinical evaluation considerations
– The acceptability of limitations in pre-market clinical data for orphan devices,
– Key considerations on the clinical evaluation of new and legacy orphan devices,
– Generating post-market clinical data for orphan devices, including PMS and PMCF.
PART B – Procedural considerations
– Guidance for notified bodies on the assessment of orphan devices,
– The role of expert panels in the context of orphan devices.
There are three appendices to this document, which include guidance on:
– OD-specific factors to include in the clinical evaluation report,
– Consideration on clinical investigations of orphan devices,
– Extrapolation of clinical data to orphan indications.
3. Scope
This document provides guidance to manufacturers and notified bodies on the clinical evaluation pursuant to the MDR of medical devices and accessories for medical devices that qualify as ‘orphan devices’ (OD) and medical devices and accessories for medical devices that have an orphan indication, within the meaning of this guidance. This guidance is relevant to devices across all risk classes as per the classification rules defined in the MDR (4).
This guidance gives particular attention to the clinical evaluation and investigation requirements stated in MDR Chapter VI and Annex XIV for these devices. Where relevant, this guidance should be read in conjunction with other MDCG guidance on the clinical investigation and evaluation of medical devices, including MDCG 2020-5, MDCG 2020-6, and MDCG 2023-7.
Custom-made devices, in-house devices, products without an intended medical purpose listed in MDR Annex XVI and in vitro diagnostic medical devices are outside the scope of this guidance.
Please note, this document gives guidance on the clinical evaluation of orphan devices which require clinical data to demonstrate conformity with GSPRs. Guidance is not provided in this document for those specific circumstances where MDR Article 61(10) applies to an orphan device.
4. Orphan device status and orphan indication
4.1. Orphan device criteria
For the purpose of this guidance, a medical device or an accessory for a medical device should be regarded as ‘orphan device’ (hereafter also referred to as ‘OD’), if it meets the following criteria:
- the device is specifically intended to benefit patients in the treatment, diagnosis, or prevention of a disease or condition that presents in not more than 12,000 individuals in the European Union per year (5); and at least one of the following criteria are met:
- there is insufficiency of available alternative options for the treatment, diagnosis, or prevention of this disease/condition, or
- the device will offer an option that will provide an expected clinical benefit compared to available alternatives or state of the art for the treatment, diagnosis, or prevention of this disease/condition, taking into account both device and patient population-specific factors.
It is important to note that the status as an orphan device does not confer market exclusivity for that device. For the sustainable development of orphan devices (and retention of legacy orphan devices), the criteria should not be interpreted so as to prevent more than one device in a given therapeutic area being designated as an OD. Similarly, the existence of an OD in a specific therapeutic area is not alone a reason to prevent a manufacturer from justifying OD status for another similar device intended for use in the same disease or condition.
4.2. Justification of orphan device status
A manufacturer who claims that his device is an OD should provide information that supports the OD status. This information should be included in any documentation submitted to a notified body or an expert panel for the purpose of determining the OD status and, eventually, in the Clinical Evaluation Report (CER), see also section 11 and Appendix A.1.
The information justifying the OD status should be based on scientific rationale addressing at least epidemiological and device-related considerations (see non-exhaustive guiding principles below). It is to be distinguished from the clinical evidence that is required for the purpose of conformity assessment.
Manufacturers and notified bodies may seek advice from the expert panels on the OD status (see section 11).
4.2.1. Epidemiology of the disease or condition
The manufacturer should provide a description of the specific disease/condition that presents in not more than 12,000 individuals in the EU per year for which the respective orphan device is intended to be used (hereafter referred to as ‘orphan population’).
The manufacturer may also justify that the device is intended to be used to benefit patients in the treatment, diagnosis, or prevention of a disease or condition that presents in a clinically valid patient sub-population (not more than 12,000 individuals in the EU per year) within a disease or condition with an annual incidence of more than 12,000 in the EU (hereafter referred to as an ‘orphan subpopulation’).
The manufacturer should provide documentation to support that it meets the epidemiological based criterion by way of population incidence estimates. It is acknowledged that documentation to support the incidence criteria will be limited in some cases. Authoritative references (e.g., from peer-reviewed medical literature and/or public health statistics) relevant to the EU population should be provided where available.
The manufacturer may consider including additional supporting data for incidence estimates, for example from national level data, health service level data or from independent clinical experts or medical society consensus statements.
Where data available to the manufacturer are limited, for example to national or regional level only, the OD status may be justified by providing an EU population incidence based on extrapolation and considering relevant factors including heterogeneity of the incidence across the EU.
In cases where the device is intended to treat, diagnose, or prevent a rare disease as defined in the EU and affecting no more than 5 in 10,000 persons in the EU (6), and where the device is expected to be used in not more than 12,000 such individuals per year, this can be accepted as sufficient justification of the epidemiological part of the criteria.
For orphan subpopulations, the manufacturer should provide information to justify the existence of a valid orphan subpopulation for the purpose of justifying OD status for a device used in a disease/condition that presents in more than 12,000 individuals per year. This can include providing a scientific rationale for why the device is only intended for use within that subpopulation and the intended use would not be appropriate for the wider population with a non-rare disease/condition.
Arbitrary limitations of use to only a subpopulation of patients to meet the incidence criteria will not be considered sufficient as it could be clinically appropriate to use the same device and therefore generate more clinical data in the remaining larger population with the non-rare disease or condition.
Factors to consider when determining if a valid and medically plausible orphan subpopulation exists include device-specific factors such as mechanism of action, and patient-specific factors that make it medically plausible that the device is for use only for that specific subpopulation of patients. For example, an OD may be intended for use in a paediatric subpopulation or in a subpopulation within a disease/condition based on device and patient factors that make it appropriate for use only in that valid subpopulation, for example based on diversity of anatomy (7).
Other patient factors may demonstrate a valid orphan subpopulation, for example, the benefit/risk of using the device may only be positive in a subpopulation of patients who are refractory to, or not medically suitable for, alternative treatments. Literature references and clinical expert statements should be provided where available to substantiate the justification.
4.2.2. Device description, insufficiency of alternatives, expected clinical benefit
The manufacturer should provide a description of the device, its intended purpose, and a scientific rationale for why the proposed intended use is considered necessary or important in the context of the management of the orphan population (or orphan subpopulation) in question, with reference to device-specific factors. If relevant, the manufacturer may choose to describe a specific indication for use in addition to, or in place of, an intended use, to assist in providing this justification. The device description should include a description of the current state of the art and alternative therapies (if any, including the relative availability of alternatives) to justify the relevance of the intended use or indication.
An explanation should be provided as to why the device would provide an expected clinical benefit compared to available alternatives or state of the art for the treatment, diagnosis or prevention of the disease/condition. Information from medical literature (for example clinical treatment guidelines) or consensus statements from clinical experts or medical societies, which may include patient representative groups, may be used to support the justification of the expected clinical benefit, for example where they detail relevant gaps in clinical management of the disease in the existing state of the art and why the therapeutic option to be provided by the proposed OD is needed. Relevant non-clinical and preliminary clinical data on the device, and/or data on similar devices, may be used in support of a statement that the OD will provide an expected clinical benefit.
4.3. Orphan indication
In addition to devices considered to have OD status based on the criteria described in section 4.1, it is recognised that a device may have a specific intended purpose/indication for an orphan population, or orphan subpopulation, where the device also has another intended purpose/indication in larger patient populations. In such cases, where duly justified, the principles outlined in this guidance for demonstrating sufficient clinical evidence may be applicable to these devices for the purposes of supporting their orphan indication only and without prejudice to the clinical evaluation requirements for other intended purpose/indications in view of their certification in accordance with the MDR. In such cases the manufacturer should justify that the intended purpose in the orphan population, or orphan subpopulation, is sufficiently different to the other intended purpose/indications, such that the clinical evidence for the non-orphan intended purpose/indication(s) is not applicable and there is an authentic challenge to generating clinical data for the orphan indication.
In the case of orphan indications, the extrapolation of (clinical) data available about the device intended for use in the non-orphan (e.g., adult) population may be of particular relevance. The appropriateness of extrapolation largely depends on
– similarity between the existing non-orphan and orphan (sub-)population characteristics;
– the quality of the available data in terms of study design, data collection, and measurement;
– and whether the extrapolated evidence constitutes valid scientific evidence.
Further considerations for extrapolation of data are provided in Appendix A.3.
PART A – Clinical Evaluation Considerations
5. The acceptability of limitations in pre-market clinical data
Having regard to the challenges to generate clinical data in the premarket phase, orphan devices may be granted market access with acceptable limitations in the amount and quality of pre-market clinical data, provided that appropriate measures are implemented, as described in this document. There must be sufficient clinical evidence to demonstrate an expected clinical benefit and that the device performs as intended with an acceptable level of safety. To address and resolve any limitations in pre-market clinical evidence as soon as possible, an adequate PMCF plan must be developed to ensure appropriate collection and generation of post-market clinical data.
As with all devices, orphan devices must meet the GSPRs that apply to it (8). For relevant GSPRs (9), there must be sufficient clinical evidence to demonstrate conformity. The manufacturer must specify and justify the level of clinical evidence necessary for demonstrating conformity with those relevant GSPRs, taking into consideration the characteristics of the device and its intended purpose (10). When specifying the level of clinical evidence necessary for an orphan device, important characteristics to consider include the clinical disease/condition being treated, the anticipated risks, the insufficiency of alternatives and unmet medical need, and the expected clinical benefit that the device offers, balanced against the anticipated risks.
An overall evaluation should be completed, to determine and justify whether the current level of clinical evidence is sufficient for the purpose of placing the orphan device on the market. In general, a limited level of pre-market clinical data is acceptable for the purpose of conformity assessments pursuant to MDR Article 52 of an orphan device if the following can be justified for the orphan device in question:
– all available non-clinical and clinical data relevant to the orphan device have been evaluated (11), and any limitations in clinical data have been identified;
– the existing non-clinical and limited clinical data is sufficient to demonstrate that the relevant GSPRs in Annex I MDR are met, that the benefit-risk ratio is acceptable, and that it is expected that the device will provide a clinical benefit taking into account the clinical condition, the state of the art, and the safety of patients;
– it is not feasible or proportionate to generate further clinical data within an acceptable time frame in the pre-market setting;
– the manufacturer has an adequate PMCF plan that, once executed, will generate clinical data in an appropriate time frame that will fully address the remaining limitations in clinical data.
– users of the device will be adequately informed (e.g. by provision of information in the IFU, SSCP (for implantable and class III devices), and/or other accompanying documentation) of the orphan status of the device, the limitations in pre-market clinical data, and instructions to users on how to report incidents, complaints, and other clinical experience to the manufacturer.
For example, an orphan device might have known limitations with respect to clinical data confirming the long-term safety or performance of the device, but there may be sufficient clinical evidence for the purposes of market access, provided that the above listed points have been adequately justified, including the development of a PMCF plan that will collect long-term data in an appropriate way.
While the general principles and methodology of the clinical evaluation process also apply for OD, the following sections will give granular detail on specific considerations for the clinical evaluation of orphan devices and its assessment by notified bodies.
6. The role of non-clinical data
‘Non-clinical data’ is understood as any relevant data that does not meet the MDR definition of clinical data (12). This includes pre-clinical data as described in the MDR (13). Non-clinical data can have a supportive role in establishing what the acceptable safety, performance and benefit-risk profile of the device will be. In addition to their role in pre-clinical evaluation, some non-clinical data may have a greater role in clinical evaluation of certain devices.
The previously discussed limitations in the ability to generate pre-market clinical data increase the importance and relevance of robust, high-quality non-clinical data. All potential sources of non-clinical data should be considered for orphan devices. If the non-clinical data provide substantial high-quality evidence to support the safety and performance of the orphan device and its expected clinical benefit, this can reduce the burden of required pre-market clinical data and can help to justify CE marking with limitations in clinical data that can be met through PMCF activities.
Useful sources of non-clinical data can include:
– Results of laboratory and animal tests;
– Data from computer modelling and simulated use testing, including software-based models, 3D printed models, and other physical models;
– Data from ex vivo studies and cadaveric studies;
– Data from similar devices (per MDCG 2020-5, section 5), for which equivalence is not demonstrated (not qualifying as clinical data per MDR);
– Information with regard to the state of the art of the technology;
– Datasets with previously collected information on patients’ health. These can be used to test the device without exposing patients, most commonly to validate software.
– Any other relevant data involving humans, which does not qualify as MDR clinical data.
The use of these data should be duly justified by providing clear explanation on their relevance with regard to the orphan device.
7. Clinical evaluation overview
The requirements for the clinical evaluation of medical devices laid down in Article 61 and Annex XIV of the MDR also apply to orphan devices. These include the following steps:
– establish and update a clinical evaluation plan;
– identify relevant clinical data and any limitations in clinical data;
– appraise all relevant clinical data;
– analyse all relevant clinical data;
– generate new or additional clinical data needed to address outstanding issues;
– document this evaluation in a clinical evaluation report; and
– update the clinical evaluation through PMCF activities.
7.1. Clinical evaluation plan
The aspects listed below should be addressed when developing the clinical evaluation plan for orphan devices, as they are important factors that need consideration when determining the acceptability of limitations in pre-market clinical data.
Disease-specific factors
– Information on the relevant disease(s)/condition(s)
- Epidemiology, including incidence supporting the OD status;
- Patient population affected by the disease or condition where the OD is intended to be used, including vulnerable populations such as minors;
- Severity of the disease/condition, including details on its morbidity and mortality;
- Factors of the disease or condition that contribute to the challenges and difficulties to generate pre-market clinical data in this population, including (if applicable) legal or ethical challenges to conducting clinical investigations in relevant vulnerable populations.
– Current state of the art in management of the disease/condition in question
- Identify and describe all relevant alternative diagnostic and/or therapeutic options (if any)
- Explain the relevant limitations (if any) in clinical management of the disease in the existing state of the art.
Device-specific factors
– Summary of supporting information to justify that the device meets the criteria for orphan device status, as described in section 4;
– Summary of pre-clinical evaluation, including relevant non-clinical data;
– Justification for acceptability of limitations in pre-market clinical data, as described in 5.
7.2. Identifying, appraising, and analysing clinical data
As with all medical devices, the clinical evaluation of orphan devices requires appropriate identification of relevant clinical data, appraisal of the quality and scientific validity of each data source, and analysis of the results and conclusions of these data sources. These steps should be followed when evaluating clinical data for the device, e.g. from previous clinical investigations and studies, previous use under derogations/compassionate use, use in the post-market setting (e.g. data from use in the EU for legacy devices, data from devices already in use outside of the EU).
Once all existing non-clinical and clinical data have been evaluated, the manufacturer should be able to determine the following, regarding their device:
– which GSPRs and clinical evidence requirements have been met, in total or in part, through existing non-clinical and clinical data,
– which requirements (if any) require additional clinical evidence, either pre-market or post-market, to fully meet these requirements.
– what are the current limitations in clinical data.
By knowing the current limitations in clinical data, the manufacturer can generate additional clinical data in a focussed manner to address those specific limitations, with objectives and endpoints that specifically aim to address those limitations. It may be acceptable for some limitations in clinical data to be addressed through specific PMCF activities.
7.3. Clinical evaluation of orphan devices which are legacy devices
A substantial group of orphan devices also qualify as legacy devices, here referred to as ‘legacy orphan devices’. Thus, in addition to the guidance in this document, the detailed guidance on clinical evidence requirements for legacy devices, including those outlined in MDCG 2020-6 and MDCG 2023-7, are directly relevant to this group of orphan devices.
The following observations should be noted regarding orphan devices which are legacy devices:
7.3.1. Sufficient clinical evidence
For orphan devices which are legacy devices, it may be justifiable to generate some or all new clinical data in the post-market phase following CE-marking under the MDR, provided that the guidance and provisions outlined in this document are appropriately followed.
7.3.2. Data from clinical investigations and legacy orphan devices
As outlined in MDCG 2023-7, the requirement for a pre-market clinical investigation specified in MDR Article 61(4) does not apply to legacy devices, provided they can otherwise demonstrate sufficient clinical data (per MDR Article 61(6)a). This clinical data can include data from prior use under the AIMDD or MDD, such as PMCF data, PMS safety data, retrospective studies of registry data, data from previous independent research, etc. Further guidance is outlined in MDCG 2020-6.
7.3.3. Clinical data from equivalent devices
Orphan devices, like all devices, may avail of the MDR provisions for equivalence, in line with the requirements laid out in MDR Article 61 and Annex XIV. In some circumstances, a manufacturer of an orphan device which is a legacy device may intend to demonstrate equivalence with a device or devices that are no longer on the EU market. Manufacturers should refer to MDCG 2020-5 and MDCG 2023-7 if considering the use of equivalence as a source of clinical data for a legacy orphan device.
7.3.4. Clinical data from off-label use
For certain legacy devices, there may be circumstances where the device has been systematically used off-label for an orphan indication by the clinical community across the EU/world for many years, to the extent that it is now considered by clinical experts as part of best clinical practice for the management of that disease or condition. As such, there may be an existing substantial body of clinical data, e.g. real-world data, supporting this indication as part of the clinical evaluation.
In some circumstances, it may not be feasible or appropriate to conduct a clinical investigation prior to extending the intended purpose for these devices, for example, where there is a lack of equipoise in the clinical community and the clinical evidence demonstrates that the off-label use is standard clinical practice and offers clinical benefit above any available alternative therapeutic options.
In such circumstances, with respect to the MDR clinical evidence requirements, it might be acceptable to consider clinical data from off-label use when considering revision or expansion of a device’s intended purpose to include this use/indication, provided that (in addition to the guidance stated in this document):
– the decision to not perform a clinical investigation is justified and compliant with relevant MDR requirements (14);
– the off-label clinical data is of sufficient amount and quality to allow clinical evaluation and notified body assessment; and
– the PMCF plan sufficiently justifies how the limitations in clinical data will be addressed through PMCF activities.
Please note, this scenario is only foreseen for exceptional cases of legacy orphan devices or orphan indications with respect to legacy devices and is not expected to apply to new orphan devices.
8. Generating pre-market clinical data for orphan devices
For ODs which are implantable devices and/or class III devices, MDR Article 61(4) requires the performance of clinical investigations, unless one of the exemptions laid down in MDR Article 61(4), (5) or (6) applies (15). Clinical investigations of orphan device can be challenging to perform due to the limited numbers of affected subjects and the scarcity of the available data. When designing a clinical investigation for orphan devices, involvement of appropriate clinical experts should be sought to ensure the study is appropriately designed to reflect the clinical needs of the target population. In addition, engagement with patients, patient associations, parents and/or caregivers of the target population may also be helpful in confirming whether the study includes patient-relevant clinical outcomes.
When designing a clinical investigation for orphan devices, potential recruitment challenges and sample size implications should be taken into account. Strategies should be considered on how best to recruit and retain patients, considering the geographical distribution and potential logistical challenges. Efforts should be made to collaborate with multiple centres where appropriate and proportionate to ensure sufficient participation and to enhance the potential for generalisability of results.
Appendix A.2 provides further considerations that should be made when designing a clinical investigation for orphan devices, with particular attention to:
– Defining the study population
– Selecting appropriate objectives and endpoints
– Choice of study design
– Choice of comparator
– Study monitoring
For the clinical evaluation of orphan indications, it may be appropriate to avail of clinical data extrapolated from the use of the device in other, non-orphan populations. Considerations on the appropriate extrapolation of these data are outlined in Appendix A.3.
9. Post market surveillance and PMCF for orphan devices
The specific characteristics of orphan devices and the limitations in pre-market clinical data make the post-market surveillance and clinical follow-up processes more important for the life-cycle evaluation of orphan devices.
If pre-market limitations in clinical data have been identified and deemed acceptable (as described previously), it is important that these limitations will be filled through well-defined and structured PMCF activities. Therefore, for orphan devices, the manufacturer must have an appropriately structured and detailed PMCF plan which is subject to notified body review in line with the conformity assessment pursuant to MDR Article 52. It should be ensured and verified that the PMCF plan is implemented and followed to completion, otherwise increased reliance on post-market clinical data collection could undermine patient safety if necessary and timely data collection does not take place.
The PMCF plan should include information about:
– All limitations in clinical data identified pre-market, that need to be addressed,
– Justification as to how the PMCF activities will address these specific limitations,
– The type of data to be generated in the post-market phase to further evaluate the clinical performance and safety of the device,
– How these data will be generated in an appropriate time frame, including projections on the numbers of patients that will be managed with the device per year, and pre-defined milestones on the periodic analysis of these data, where appropriate.
This may include data collected from PMCF investigation(s), registries or other clinical data and clinically relevant information in the post-market setting, including real-world data, as described below. It should be noted that PMCF investigations and registries are not necessarily both required.
Depending on the limitations in pre-market clinical data and the reliance on PMCF to address these limitations, it may be appropriate for specific conditions or provisions to be defined by the notified body for the certification of these devices (16). For example, it may be appropriate for specific PMCF activities and milestones to be required by the notified body as specific provisions of certification of the device.
When developing a PMCF plan, due consideration needs to be given with respect to the potential challenges that may be faced during execution of the PMCF plan, which may require adjustment of the expected time frame and milestones for the collection of post-market clinical data. To this end, continued structured dialogue between the manufacturer and the notified body may be appropriate to discuss any challenges faced with respect to delivery of the PMCF plan.
9.1. PMCF and benefit-risk determination
The data generated from PMCF is important to enable the continued assessment of the benefit-risk profile of orphan devices with a higher degree of certainty. Thus, it will be important to develop a detailed risk management plan adapted to the unique characteristics of the orphan device, its intended use, and the limitations in pre-market clinical evidence. This plan should outline how risks will be monitored, assessed, and mitigated throughout the PMCF, and outline how the manufacturer will take appropriate action where this data raises new concerns regarding the safety or performance of these devices.
9.2. PMCF investigations
The considerations outlined in Appendix A.2 on clinical investigations with orphan devices also apply for PMCF investigations and should be taken into account. Furthermore, in addition to the general requirements and expectations for PMCF investigations, particular attention should be made for PMCF investigations with orphan devices in the following areas:
– Consider any possible condition(s)/specific provision(s) provided by the notified body in the certification and their impact on the PMCF investigation.
– Consider how the proposed study will address any limitations in clinical evidence identified in the pre-market phase.
– Orphan devices may require long-term follow-up studies to assess the durability of the device’s benefits and to identify any potential long-term safety concerns. While pre-market studies may include limited follow-up periods, PMCF investigations should allow for continuous monitoring and evaluation of orphan devices over extended periods of time. Long-term follow-up is especially important in diseases or conditions with slow progression or where the benefits of the device may manifest over an extended period.
– Where possible, for the duration of recruitment into a PMCF investigation, the manufacturer should plan to enrol a representative majority (e.g. greater than 90% where feasible (17)) of patients exposed to the device in each investigational site. This is particularly important for devices that carry significant risks (i.e. high residual risks or risks of causing serious adverse events).
– In addition to collecting objective clinical data reflecting safety and performance, PMCF studies may also capture, as secondary endpoints, real-world usability data from the user/healthcare professional perspective and/or considering need for additional usability studies in case pre-market data are incomplete due to not being able to replicate the real-life conditions of use (see also section 9.4).
9.3. Registries
In relation to orphan devices, registries are considered as a valuable tool in building a broad and comprehensive knowledge base for these often-heterogeneous diseases. As such, it is recommended that orphan devices are enrolled in registries as part of PMCF plan where appropriate and feasible.
As part of PMCF, the manufacturer should identify and/or support the development of suitable registries for collecting sufficiently representative (e.g., greater 90% where feasible) data on patients with the disease/condition as well on those receiving the specific orphan device, with the aim of comparing outcomes in patients treated and not treated with the OD across Europe. Where available and suitable, manufacturers are strongly encouraged to use registries established and governed by national bodies or speciality medical associations.
The manufacturer should substantiate and justify why chosen registries are suitable for providing this data and how they will address the identified limitations in pre-market clinical data. The manufacturer should use available guidance and suitable methodologies to demonstrate sufficient access to, and quality of, the data within the registries, for the purposes of confirming the safety and performance of the device throughout the device’s life cycle.
9.4. Other post-market clinical data and post-market surveillance
In the post-market setting, clinical data can be collected from sources other than through PMCF clinical investigations. These sources are often referred to as ‘real world data’ and can be used to generate ‘real world evidence’. This clinical data is collected in the post-market setting (e.g., during PMS or certain PMCF activities like registries), during the routine use of the device in clinical practice. As with all devices, manufacturers of orphan devices must have an appropriate PMS system in place (18). In addition to the PMCF activities discussed above, the clinical data collected from PMS should be evaluated as appropriate, as part of the life-cycle evaluation of these devices.
The target population of an orphan device can be geographically, ethnically, and physiologically diverse. The evaluation of real-world data can help to detect rare complications and understand factors such as ethnicity, which may affect the clinical performance of the device. This data is of particular relevance to legacy orphan devices and should be considered where available, provided that the clinical data is of sufficient quality for the purpose of clinical evaluation and assessment.
PART B – Procedural Considerations
10. Notified body activities and responsibilities
10.1. Notified body activities prior to the certification
The OD status of the device should be checked by the notified body as early as possible, for example as part of structured dialogue (19) before or during initial conformity assessment activities. This should be based on the justification and information provided by the manufacturer (see section 4.2) and, if applicable, advice provided by an expert panel to the notified body or the manufacturer (see section 11).
When the orphan device status is established, the technical documentation should be assessed following the same principles as for non-orphan medical devices. However, when assessing the manufacturer’s clinical evaluation plan and clinical evaluation report (CER), the product reviewers/clinical experts should consider the aspects addressed in this guidance, including the acceptability of limited pre-market clinical data and appropriate PMCF activities to generate additional clinical data.
The notified body’s assessment of the CER should address the information supporting the orphan device status and, where applicable, the rationale for accepting limitations in the pre-market clinical data and the activities proposed by the manufacturer in its PMS plan and PMCF plan to obtain the necessary additional clinical data.
10.2. Specific conditions/provisions for certification
The MDCG acknowledged in its position paper MDCG 2022-14, point 17 that the use of certificates with conditions will contribute to increasing the necessary flexibility to apply the reinforced clinical evidence requirements to devices that have a demonstrable track record of safety. Orphan devices for which the pre-market clinical evidence is deemed sufficient but needs to be completed or confirmed through PMCF, are a good example where notified bodies can make use of the possibility to issue certificates with specific conditions or provisions.
Specific conditions or provisions (20) may consist, for example, in requiring the manufacturer:
– to conduct defined PMS or PMCF activities (21) within a specified period of time to generate additional clinical data (see section 9),
– to adequately inform users of the device of the orphan status of the device, the limitations in pre-market clinical data, and instructions to users on how to report incidents, complaints, and other clinical experience to the manufacturer, e.g. by provision of information in the IFU, SSCP (for implantable and class III devices) and/or other accompanying documentation (see section 5).
10.3. Surveillance by the notified body
The notified body should consider PMS data, in particular the main findings from PMCF as part of the agreed surveillance activities and PSUR evaluation pursuant to MDR Article 86, and verify whether the device’s benefit-risk profile continues to support the placing of the device on the market. As part of their surveillance activities and post-certification monitoring, notified bodies need to monitor compliance with any conditions/provisions that are binding for the manufacturer and associated with the certification decision, such as updates to clinical data at defined intervals (22). Where applicable, especially if listed as part of the conditions for certification, the notified body also needs to review the clinical evaluation that the manufacturer has updated based on its PMS, PMCF (23).
When the conditions/provisions on the certificates are not fulfilled/met by the manufacturer, the notified body should consider the impact thereof on the certificate’s validity, as specified in their procedures. Not fulfilling the conditions/provisions could ultimately lead to suspension or withdrawal of the certificate.
11. Involvement of expert panels: advice on orphan device status and clinical evidence
While it rests with the manufacturer to demonstrate that its device meets the criteria for orphan device status, the expert panels established in accordance with MDR Article 106 (24) may be requested to provide advice on the orphan device status and the clinical data needed for the clinical evaluation.
11.1. Consultation of expert panel
The consultation of an expert panel in relation to an orphan device described in this section is optional/voluntary and independent of the clinical evaluation consultation procedure (CECP) provided for in MDR Article 54(1).
The following paragraphs address different scenarios for the consultation of an expert panel depending on the state of advancement of the device development or the conformity assessment. To improve predictability with respect to clinical evidence requirements, a manufacturer or notified body may seek independent advice from an expert panel as to whether its device meets the criteria of an orphan device as early as possible.
11.1.1. Early scientific advice pursuant to MDR Article 61(2)
Article 61(2) MDR provides the possibility for a manufacturer, prior to its clinical evaluation and/or investigation, to consult an expert panel with the aim of reviewing the manufacturer’s intended clinical development strategy and proposals for clinical investigation. The scope of MDR Article 61(2) is limited to class III devices and class IIb active devices intended to administer and/or remove a medicinal product.
The orphan device status will influence the expected level of pre-market clinical evidence, notably the justification for limitations in the pre-market clinical evidence and an acceptable level of pre-market clinical uncertainty (see sections 3-7 of this guidance). It is therefore recommended that manufacturers of devices that fall within the scope of MDR Article 61(2) and that may qualify as OD consult an expert panel on their intended clinical development strategy in accordance with MDR Article 61(2). This advice procedure may be particularly useful for new orphan devices.
Where a request for such an early scientific advice concerns an orphan device, the expert panel will, as a necessary first step, assess the manufacturer’s justification regarding the orphan device status. In a second step, the expert panel will review the manufacturer’s intended clinical development strategy and proposals for clinical investigation, which – for orphan devices – may particularly include proposals for PMCF. Pursuant to MDR Article 61(2), the manufacturer shall give due consideration to the expert panel’s views on the orphan device status and on its clinical development strategy and document this consideration in its clinical evaluation report.
11.1.2. Advice in cases where the clinical evaluation is in an advanced stage or completed
The early scientific advice pursuant to MDR Article 61(2) would be too late for manufacturers who have already drawn up their clinical evaluation report or are in an advanced stage with their clinical evaluation. Having regard to the deadline for lodging applications for conformity assessment by 26 May 2024 in accordance with MDR Article 120(3c), point € (25), this will be the case for many legacy devices, but it is not limited to them.
In those cases, an expert panel’s advice regarding the orphan device status and regarding the clinical data required for the clinical evaluation of a device may be requested by a notified body in accordance with MDR Article 106(11) in the framework of an ongoing conformity assessment procedure (see below point (a)).
In exceptional cases the manufacturer may request advice from an expert panel on the orphan device status and the clinical data required for its clinical evaluation, even though the clinical evaluation is in an advanced stage or already completed (see below point (b)).
The scope of the expert panel advice will depend on the request made by the notified body or the manufacturer: it may concern only the OD status, or it may also concern the clinical data required for the manufacturer’s clinical evaluation, including any justification regarding limited clinical data, the acceptability of clinical uncertainty and proposed post-market clinical follow-up activities.
(a) Advice requested by a notified body
The notified body involved in the conformity assessment of a device for which the manufacturer claims an orphan device status may seek advice from an expert panel in accordance with MDR Article 106(11).
Before submitting such a request, the notified body should consult the manufacturer, for example to inform them of their plan to request advice from the expert panel and where appropriate to give the manufacturer the opportunity to provide input into the request. Having regard to the limited capacity of the expert panels, notified bodies are advised to reach out to the EMA expert panel secretariat as early as possible to include an envisaged request for advice in the expert panels’ planning.
The request for advice from the notified body may concern the orphan device status and possibly also the clinical data required for the manufacturer’s clinical evaluation, including any justification provided by the manufacturer regarding limited clinical data, the acceptability of clinical uncertainty and proposed post-market clinical follow-up activities. For that purpose, the notified body should put forward to the expert panel specific questions for which it seeks the panel’s advice. Those questions should be based on a preliminary analysis of the clinical evaluation provided by the manufacturer.
To ensure a consistent approach in the conformity assessment of orphan devices, the MDCG encourages notified bodies to make use of this consultation , particularly in those cases where the notified body does not agree with the manufacturer’s claim that the device qualifies as an orphan device, or if it is uncertain in this regard, unless an expert panel has already provided advice on the orphan device status to the manufacturer.
In those cases, the notified body should consult the expert panel at an early stage of the conformity assessment procedure, e.g. in the application review phase.
Where the requested advice concerns the clinical evaluation, the notified body should determine the timing of the consultation in agreement with the manufacturer depending on how it fits best in the overall conformity assessment procedure.
The notified body will need to include the expert panel’s considerations in its clinical evaluation assessment report. If the notified body has a different view than the expert panel, it should give reasons for such divergent views in its clinical evaluation assessment report.
(b) Advice requested by the manufacturer
As an extraordinary measure during the MDR transitional period, which ends on 31 December 2027 or 31 December 2028 depending on the device’s risk class, a manufacturer who has already completed its clinical evaluation, or is in an advanced stage with it, may request advice from an expert panel on the orphan device status and possibly also on the clinical data required for the clinical evaluation, provided that the request for an expert panel advice does not interfere with the assessment by the notified body.
To avoid any overlap with the technical documentation assessment by the notified body, a manufacturer should only request advice from an expert panel if it will be able to update its clinical evaluation report taking into consideration the expert panel’s views, before the notified body assesses the manufacturer’s clinical evaluation (26). The manufacturer’s clinical evaluation plan or its draft clinical evaluation report could be suitable documents to be submitted with the request. The manufacturer should inform the notified body about the request and about any advice provided by the expert panel, for example, in its application for conformity assessment. The manufacturer should make the expert panel advice available to the notified body, for example as an annex to the clinical evaluation report.
If the manufacturer intends to request advice from an expert panel after it has already lodged an application to a notified body, the manufacturer and notified body should agree that the expert panel consultation does not interfere with the notified body’s assessment. If this cannot be ensured, a consultation of the expert panel should be left to the notified body pursuant to MDR Article 106(11).
11.2. Timelines for expert panel’s advice
The MDR does not set a deadline for expert panels to provide their advice. However, Table 2 of the Commission Implementing Decision (EU) 2019/1396 as regards the designation of expert panels in the field of medical devices (27) lays down the maximum number of days for which experts may be remunerated for certain tasks, such as scientific advice, distinguishing between simple matters, complex matters, and very complex matters.
11.2.1. Early scientific advice pursuant to MDR Article 61(2)
For early scientific advice under MDR Article 61(2), EMA’s expert panels’ secretariat has set up a process consisting of different steps which aims to ensure that the applicant manufacturer submits an appropriately prepared request and that the expert panel provide its advice in a timely manner.
11.2.2. Advice in cases where the clinical evaluation is in an advanced stage or completed
Where the expert panel advice is limited to the question whether a device meets the criteria of an orphan device, the expert panel will endeavour to provide its advice within 60 days.
Where the expert panel advice concerns both the orphan device status and the clinical data used by the manufacturer for its clinical evaluation, the expert panel will endeavour to provide its advice within 90 days provided it is simple advice. The advice should clearly distinguish between the expert panel’s views on the orphan device status and its views on clinical data related aspects.
If considered appropriate by the expert panel, it can provide its advice in two steps: first advice on the orphan device status (to be provided within 60 days) and second advice on the clinical data used by the manufacturer for its clinical evaluation (to be provided within 60 days from the date of the issuance of the advice on the orphan device status, if it is simple advice as mentioned before).
11.3. Relationship with CECP
The CECP provided for in MDR Article 54(1) may apply to a device for which the manufacturer or a notified body have requested expert panel advice in accordance with this guidance. In such a case, the expert panel advice and how it has been taken into consideration by the manufacturer or the notified body should be reflected in the notified body’s clinical evaluation assessment report that is submitted for CECP. The notified body should indicate in their CECP submission if the device has been subject to a voluntary advice procedure (28).
Appendices
A.1. Clinical Evaluation Report
A.1.1. OD-specific information to be included in the CER
As part of the general requirements regarding the CER for all devices, the manufacturer should ensure that the CER includes summary descriptions of the orphan device-specific considerations laid out in this guidance, including:
– summary of how the device meets the criteria for orphan device status, per section 4;
– summary of any identified limitations in clinical data and residual risks, including a description of how these were identified;
– acceptability of these limitations in clinical data and residual risks, per section 5, with particular justification that:
- all available non-clinical and clinical data relevant to the orphan device have been evaluated (29), and any limitations in clinical data have been identified;
- the existing non-clinical and limited clinical data is sufficient to demonstrate that the relevant GSPRs in Annex I MDR are met, that the benefit-risk ratio is acceptable, and that it is expected that the device will provide a clinical benefit taking into account the clinical condition, the state of the art, and the safety of patients;
- it is not feasible or proportionate to generate further clinical data within an acceptable time frame in the pre-market setting;
- the manufacturer has an adequate PMCF plan that, once executed, will generate clinical data in an appropriate timeframe that will fully address the remaining limitations in clinical data.
- users of the device will be adequately informed (e.g. by provision of information in the IFU, SSCP (for implantable and class III devices), and/or other accompanying documentation) of the orphan status of the device, the limitations in pre-market clinical data, and instructions to users on how to report incidents, complaints, and other clinical experience to the manufacturer.
– summary of the applicable non-clinical data that was evaluated as part of clinical evaluation planning as outlined in section 6;
– summary of pre-market clinical data that have been identified and evaluated, including any clinical investigations, with due regard to sections 7 and 8 and Appendix A.2;
– a clear, stringent, and detailed PMCF plan (30) with due regard to section 9, including:
- summary of the risk management plan as described in section 9.1.
- description of the type and quality of data that needs to be generated in the post- market phase in order to further evaluate the clinical performance and clinical safety of the device and address identified limitations in clinical data;
- description on how the manufacturer plans to generate these data in an appropriate, timely manner;
- projections on the number of patients that will be managed with the device per year in the EU;
- a summary of planned PMCF activities including, as applicable, PMCF investigations and registries in the EU and globally for this device;
- for orphan devices that carry significant risks (i.e. significant residual risks and/or high risk of causing serious adverse events), confirmation that the manufacturer will prospectively enrol a representative majority (e.g. greater than 90% if feasible) of patients into PMCF activities including PMCF investigations and/or registries;
– summary of any interactions with expert panels as described in section 11;
– a plan to update the clinical evaluation report at pre-defined intervals as appropriate, based on the PMCF plan, and whenever new information becomes available that may change the benefit-risk profile of the device.
A.1.2. Post-market updates to the CER
As part of updates to the CER in the post-market setting, the following OD-specific information should be highlighted and kept up to date as necessary, based on latest available information:
– up-to-date information with respect to the orphan device status, per section 4,
– total product sales in the EU and worldwide,
– summary of state of the art, highlighting any changes to available alternatives (if any),
– updates on clinical evidence including any changes to limitations in clinical data,
– summary of up-to-date determination of benefit-risk ratio,
– summary report of ongoing PMCF activities and their progress towards addressing limitations in pre-market clinical data.
A.2. Considerations for Clinical Investigations of Orphan Devices
A.2.1. Introduction
The purpose of this section is to provide illustrative examples of different approaches and considerations that device developers could consider when designing a clinical investigation of an orphan device. It is not intended to be an exhaustive or prescriptive list of requirements with respect to study characteristics. As with all clinical investigations, it is expected that clinical investigations of orphan devices will be designed and conducted in compliance with the relevant MDR requirements and in line with relevant good clinical practice principles, including an appropriate level of engagement with stakeholder groups such as clinicians, users, and patient representatives. For more guidance on clinical investigations, please also refer to MDCG 2021-6 rev. 1, MDCG 2024-3, and MDCG 2024-5.
A.2.2. Defining the study population
Careful definition of the study population is key. For orphan devices the target population is small (i.e. not more than 12,000 individuals per year) and may be vulnerable, for example infants and children. The fact that rare diseases/conditions usually affect patients from birth makes circumstances even more complex.
To this point, inclusion and exclusion criteria should be wide enough to enrol the maximum number of target population without being too general in the sense that it may introduce too much variability (i.e., heterogeneity or ’noise‘) and obscure the clinical investigation results. Many orphan devices are used in vulnerable populations, and thus it is anticipated and expected that clinical investigations may include these vulnerable populations, where appropriate. In these cases, the requirements set out in MDR Articles 64 – 68 relating to inclusion of vulnerable populations must be met.
A.2.3. Objectives
For many orphan devices, it may be suitable for the primary objectives of their pre-market clinical investigation(s) to focus on assessment of short- to medium-term clinical benefit, patient safety, and benefit-risk ratio. Additional objectives should focus on the short- to medium-term performance and technical success of the procedure and device. For PMCF investigations, the objectives should aim to evaluate the overall safety, performance, and clinical benefit of the device throughout its life cycle, with a suitable focus on long-term endpoints.
A.2.4. Selection of endpoints
Clinical performance endpoint(s) should be predefined on the basis of relevant indicators to assess the clinical outcome, safety, and clinical benefit. The appropriateness and relevance of the chosen endpoints should be clearly justified in the clinical investigation plan (CIP), to help regulatory authorities, notified bodies, and expert panels (as applicable) understand and accept these endpoints. Continuous monitoring of safety through regular reporting serious adverse events is a key endpoint throughout the evaluation program. Secondary endpoints can also be included to further establish the overall clinical benefit of the device.
Although disease-specific clinical endpoints remain the standard, such endpoints may not be sufficiently established, understood, or validated in the clinical setting for certain conditions involving orphan devices. In this regard, appropriately validated surrogate endpoints can be considered, if justified. In these cases, relevant disease-specific clinical endpoints should subsequently be investigated, where possible, in the post-market setting, e.g., in PMCF investigations.
When selecting endpoints to investigate clinical benefit, it is important to identify and consider the priorities and unmet medical needs from the perspective of the patient. To that end, patient-reported outcomes (PROs) and other patient-centric measures can be considered for inclusion as secondary/exploratory outcomes and endpoints. These can help to assess the impact of the device on the quality of life and daily functioning of the patient. However, it is acknowledged that, by their nature, PROs can be vulnerable to bias and confounding factors, and so, they should only be relied upon as primary endpoints for evaluating clinical benefit in exceptional circumstances where it can be justified that other, objective clinical performance outcomes and endpoints cannot be collected in the target population, and where the study also includes adequate, objective safety endpoints. When considering the inclusion of PROs and other clinically relevant endpoints, input from independent patients’ representatives should be sought, if available and appropriate.
A.2.5. Study design
While randomized controlled trials (RCT) are often considered the preferred study design for clinical investigations of medical devices, this study design may present challenges for orphan devices. These can include ethical challenges – for example, where there are no alternatives or where available alternatives are not considered to offer similar patient benefit (lack of equipoise) – or practical challenges – for example where the device is intended for very small patient populations, randomising some of them to a control arm may severely inhibit the ability to collect sufficiently powered data in a timely manner. In these cases, less commonly used methodological approaches may be acceptable if well justified.
Many study designs may be suitable for the clinical investigation of an orphan device. The choice of design should be carefully considered in terms of its strengths and limitations (e.g. vulnerability to bias or confounders) and its ability to address any ethical and practical challenges, and should be justified on a case-by-case basis. Illustrative examples of alternative study designs which may be suitable include cross-over designs, adaptive designs, and sequential designs.
A.2.5.1. Cross-over designs
Within a cross-over investigation each participant receives two treatments (i.e. intervention and comparator) in a random order and acts as their own control, with a “wash out” period in between. For some devices, a crossover design may be appropriate in some cases, as it will reduce the risk of confounding while also reducing the number of participants needed.
A.2.5.2. Adaptive and sequential designs
Adaptive and sequential designs are based on interim analyses planned to be carried out in the course of the clinical investigation.
Adaptive investigations may allow for potential changes in several parameters (e.g. sample size requirements, randomization ratios, number of analyses) as the study progresses. However, care should be taken to ensure the integrity of the investigation is not compromised as a consequence of excessive or unnecessary adaptations. Where possible, all anticipated or potential adaptations of the investigation should be described in the clinical investigation plan, based on anticipated results of the planned interim analyses.
For certain high-risk devices, a stepwise approach within an adaptive study design, may be appropriate and could help to assess critical but uncertain aspects such as those related to patient recruitment, methods used and variables studied, follow-up visits, and investigator and site qualifications.
Sequential designs are mainly based on interim analyses of the study’s primary endpoints, and unlike adaptive clinical investigations, no adaptation of parameters is allowed. Instead, the stepwise methodology allows for continued, periodic analyses after each predefined group/cohort/stratum of patients reach their outcome, thus allowing for interim opportunities to determine whether there is sufficient evidence of clinical benefit or lack thereof.
Although adaptive and sequential methodologies can provide more flexibility, it is important to note that this approach can be vulnerable to type I (false positive) error. This potential for error should be considered when evaluating clinical data generated from these study designs.
Where appropriate, interim analyses may be enough to support the initial clinical evaluation for conformity assessment. In this event, where possible and where there still is equipoise, the study should continue to completion as planned in the CIP, to ensure continued collection of clinical data and to ensure appropriate long-term follow-up for the study participants.
A.2.5.3. Other study designs
Other potentially suitable pre-market clinical investigation designs may include:
– Prospective observational studies of all patients exposed to the device (e.g. single arm with outcomes reported on all consecutive patients),
– Comparative studies with concurrent matched control subjects,
– Comparative studies with historical controls (if appropriate and justifiable, for example where the OD is intended for a life-threatening disease and there are no alternatives).
Within the PMCF plan, manufacturers should consider the following clinical investigation designs where appropriate:
– RCTs comparing the OD with state of the art, with appropriate blinding (e.g., double- or single-blinding, and/or blinded determination of clinical endpoints),
– Unblinded, open-label RCTs,
– Additional prospective observational cohort studies, with concurrent matched controls,
– Registry-based RCTs (a.k.a. nested trials and ‘simple RCTs’) from a suitable registry.
The choice of study design needs to be justified and appropriately documented (e.g., in the CEP, CER, and/or PMCF plan), with acknowledgement to the ethical and practical challenges for choice of study design. Consideration needs to be given with respect to minimisation of bias, representativeness of the study, and transparency of the study findings.
A.2.6. Statistical considerations and Bayesian approaches
For many clinical investigations of orphan devices, frequentist approaches to statistical analysis can be particularly challenging, as patient recruitment from an orphan (sub)population may make inferential statistical analysis unachievable or inappropriate. In such cases, descriptive statistical methods, or in some situations, alternative methods to frequentist approaches such as Bayesian approaches, may be more appropriate.
Bayesian approaches cover a very broad range of possibilities. In Bayesian analyses and hybrid Bayesian analyses, probability statements (e.g., “the probability that the experimental treatment is effective”) are made on the basis of accumulated data combined with prior (existing) data. In this sense, Bayesian approaches can also be considered adaptive.
Use of an external control, including historical control data from sources such as prior clinical investigations or ongoing registries, might be a viable approach too if it helps to improve the interpretability of the clinical investigation data. Care should be taken so that only patients with the same condition or disease and comparable clinical and demographic characteristics are used as controls.
While the use of prior data is not common to Bayesian applications, the expression of belief about prior data is however different in this framework. Belief about prior data (e.g. from literature, experts, registries) is expressed in the form of a plausible distribution. Observed data from the study are then collected and when combined with the belief about the prior data/evidence, an updated prior, known as the posterior belief (distribution) is determined. In this way, accumulated observed data combined with historical knowledge and expert opinion forms a basis for continued evaluation.
A potential advantage of using Bayesian approaches is avoidance of the constraints of the type I error which are associated with larger sample sizes. However, the absence of a type I error, does not mean that Bayesian approaches are free from other decision errors, and these should be considered. The flexibility to form a prior which expresses more or less uncertainty about prior belief can impact the sample size requirements.
Alternatively, a ‘hybrid Bayesian’ approach can be considered, which allows expression of uncertainty in the historical data, while allowing use of the frequentist approach. The justification for expression of prior belief around existing data should be rigorously supported with clinical and statistical evidence as this can have a meaningful impact on the conclusions of the study results. Where statements such as “The probability that device A has a higher response rate than device B” are used, it will be important to ensure the differences are clinically meaningful.
Other approaches may also exist which are less commonly used, for example, decision theoretic, value of information, and meta-analytic (including Bayesian evidence synthesis) approaches could be used.
However, extreme care should be exercised to ensure the methodologies are pre-specified and the clinical interpretation is clear, to avoid data driven approaches which may increase the chance of a false conclusion.
A.2.7. Choice of comparator/control
In general, a concurrent active comparator is the preferred option but may be difficult to achieve given the large number of rare diseases or conditions for which no alternative option is currently available.
In some cases, clinical data from open-label studies without control or with historical controls might be acceptable but must be well justified in terms of choice of study design; these kinds of studies are primarily intended for patients for whom there is no clinical ‘equipoise’.
Sham or no comparator can be considered but might be problematic given that those assigned to the control group may have no direct benefit. In such an event, subjects in the control arm must receive the state of the art in management of their condition – i.e., they must receive care at least to the same level as the care they would receive in normal clinical practice.
A.2.8. Monitoring
In general, appropriate monitoring by the sponsor should aim to ensure adherence to study protocols and regulatory requirements. In addition, independent monitoring (e.g., by Data Safety Monitoring Boards (DSMBs) and/or Clinical Events Committees (CECs)) should be included as appropriate to confirm that acceptable levels of safety for study participants is maintained throughout the conduct of the study.
Some statistical methods (31) can assist in the real time monitoring of severe adverse events associated to medical devices. These can be well suited for small-sized investigations and allow to re-estimate the frequency of specific emerging risks in an ongoing process (after each inclusion or group of inclusions) and allow to set stopping rules in clinical investigations by anticipating situations where the risk(s) would exceed an acceptable incidence or frequency threshold.
A.3. Extrapolation of clinical data to orphan indications
A.3.1. Key aspects
Devices that are intended by the manufacturer to be used for orphan populations or indications, as well as in non-orphan populations, may have clinical data from use of the device in the ‘other population/indication’. In some circumstances, it may be appropriate to extrapolate these clinical data from other population(s)/indication(s), for the purposes of clinical evaluation of the intended use in the orphan population/indication.
The appropriateness of this extrapolation of clinical data should be considered on a case-by-case basis, and will depend on several factors, including the characteristics of the device, the existing knowledge of the device in the non-orphan population/indication, what is known or can be extrapolated about the device to the intended orphan population/indication, and the underlying disease or condition being treated.
The extent to which extrapolated clinical data can be relied upon will vary from device to device. In general, it is anticipated that extrapolated clinical data could be combined with other clinical data to provide sufficient clinical evidence for the purpose of approval of the orphan indication (so-called ‘partial extrapolation’). Less commonly, it may be appropriate for the extrapolated clinical data to provide sufficient clinical evidence for the orphan indication without requiring additional clinical data (so-called ‘full extrapolation’). In both cases, the appropriateness of this extrapolation will need to be evaluated and justified by the manufacturer and assessed by the notified body. The notified body may consider the extrapolated data alongside any other existing clinical and non-clinical data and the PMCF plan. The PMCF plan should build on the existing data, to evaluate the safety and performance of the device throughout the device’s lifecycle for its orphan indication/intended purpose.
Partial Extrapolation: Extrapolated clinical data are combined via a suitable statistical model or methodology with other sources of clinical and non-clinical data, to provide sufficient clinical evidence to support the orphan indication/intended purpose. The construction of such a statistical model is anticipated to require the availability of measured variables that will help connect the available outcomes to the outcomes of intended orphan (sub)population/indication. If the necessary variables are not available in the data sources, partial extrapolation may not be appropriate. If the model is determined to be appropriate, then the inferences obtained from it may be used to support the orphan device indication.
Full extrapolation: Extrapolated clinical data are used as the main or sole source of clinical data (i.e. a complete substitute) to provide sufficient clinical evidence to support the orphan indication/intended purpose. Minimal to no additional clinical data are needed for the purpose of initial certification of an orphan indication/intended purpose.
The appropriateness of extrapolation of clinical data from use in other (sub)populations/indications depends on three main factors:
– the relevance of the data, including the similarity of characteristics between the proposed orphan subpopulations/indications, and the others from which the data were collected;
– the quality of the data in terms of scientific validity (32) and suitability; and
– the extent to which the data can provide sufficient clinical evidence to assess the safety, performance and expected clinical benefit for the orphan indication.
A.3.2. Decision process for considering extrapolation
To determine the suitability of clinical data for extrapolation, the following steps should be followed and documented in the clinical evaluation report.
I. Suitability for extrapolation of clinical data
Are the clinical data relevant to the orphan (sub)population(s)/indication?
Clinical data can be considered relevant for the purposes of data extrapolation if it is justified that:
(a) the indication(s) for the device is the same, but in a different population;
(b) the endpoint(s) or outcome(s) that has been measured in the non-orphan population is the same or similar as would be measured as an endpoint when used in the intended orphan (sub-)population; and/or
(c) the clinical data from the non-orphan population provide validated surrogate endpoints that are expected to be relevant to the orphan population.
- In this case, a reliable and valid model might be used to predict the endpoint for the orphan population using clinical data from the other population, e.g., a validated surrogate endpoint in the available data set(s) that has been shown to predict a different, longer-term endpoint of interest.
– If any of I(a)-I(c) is TRUE → potentially suitable for extrapolation; proceed to II.
– If NOT → unsuitable; do NOT extrapolate
II. Determining the extent of extrapolation
Do the characteristics of the device, patients, or diseases/conditions differ between the orphan and other (sub)populations/indications to an extent that the expected safety and/or performance of the device could be impacted in a clinically meaningful way?
In particular, are there clinically meaningful differences with respect to:
(a) the intended location and/or duration of use?
(b) characteristics of the device (e.g. size, morphology, indications)?
(c) patient characteristics?
- e.g., characteristics that are unique to the orphan (sub)population/indication that may impact the device’s expected safety/performance. Some devices might require special considerations relevant to only a particular subgroup of patients.
For example, special considerations may be needed with respect to difference in:- Pharmacokinetics/pharmacodynamics
- Risks related to the duration of exposure (e.g., differences in long-term toxicity)
- Age, sex, ethnicity, or body size
- For paediatric populations, potential impact of the device on physiological changes in a growing child and vice versa
- e.g., characteristics that are unique to the orphan (sub)population/indication that may impact the device’s expected safety/performance. Some devices might require special considerations relevant to only a particular subgroup of patients.
(d) disease characteristics between the two populations/indications?
(e) any other relevant differences or factors?
Other factors that may limit extrapolation include:
- insufficient knowledge of the disease or condition in the intended (sub)population,
- no suitable statistical methodology has been identified for the purposes of partial extrapolation and combination with other clinical data, e.g., the expected differences between the two (sub)populations/indications,
- the clinical data was obtained from off-label use or misuse within the non-orphan population (i.e., use in the non-orphan population is outside the intended purpose),
- the state of the art has changed since the creation of the data to such an extent that historical data would likely be different to new prospectively collected data,
- specific applicable regulatory requirements, such as the necessity for mandatory clinical investigations for certain devices (per MDR Article 61(4), see MDCG 2023-7).
– If II(a)-II(e) are ALL answered “NO” → potentially suitable for FULL EXTRAPOLATION;
appraise and analyse the clinical data (33) and proceed to III.
– If any of II(a)-II(e) answered “YES” → potentially suitable for PARTIAL EXTRAPOLATION;
appraise and analyse the clinical data2 and proceed to IV.
III. Suitability for full extrapolation
Are the extrapolated clinical data of sufficient amount and quality to provide sufficient clinical evidence for the evaluation of the device’s safety, performance, and clinical benefit for the orphan indication?
– If “YES” → likely suitable for FULL EXTRAPOLATION;
conduct clinical evaluation accordingly (34).
– If “NO” → potentially suitable for PARTIAL EXTRAPOLATION;
Proceed to IV.
IV. Suitability for partial extrapolation
Despite any differences or limitations identified, are the extrapolated clinical data suitable (2) for use as part of the clinical evidence for the orphan indication?
– If “YES” → likely suitable for PARTIAL EXTRAPOLATION;
conduct clinical evaluation accordingly (3).
– If “NO” → do NOT extrapolate
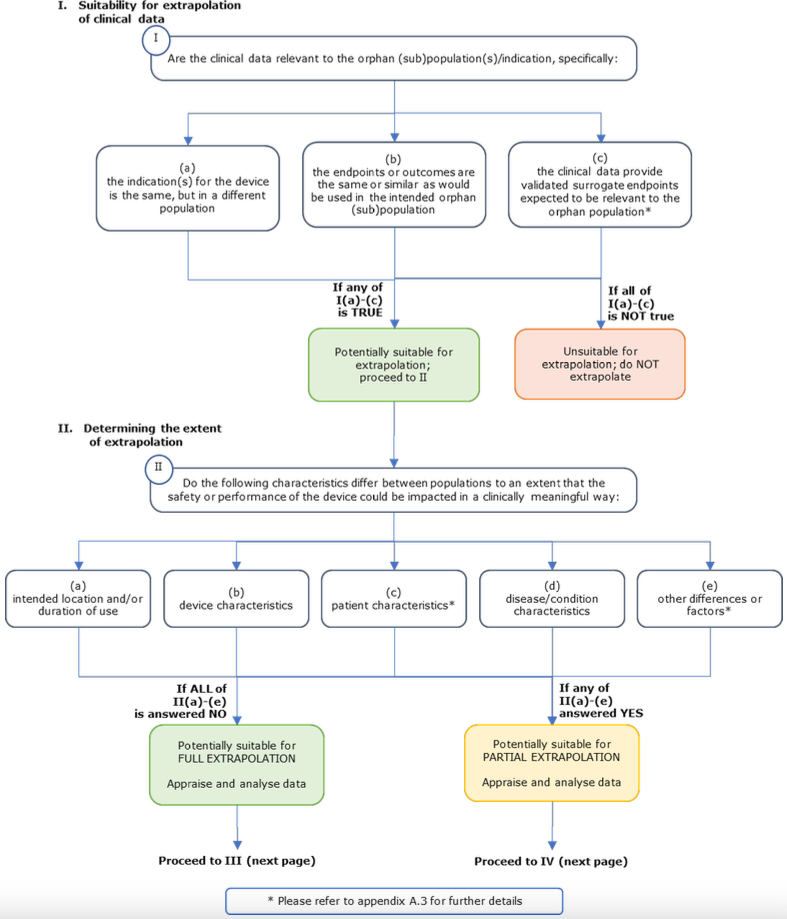
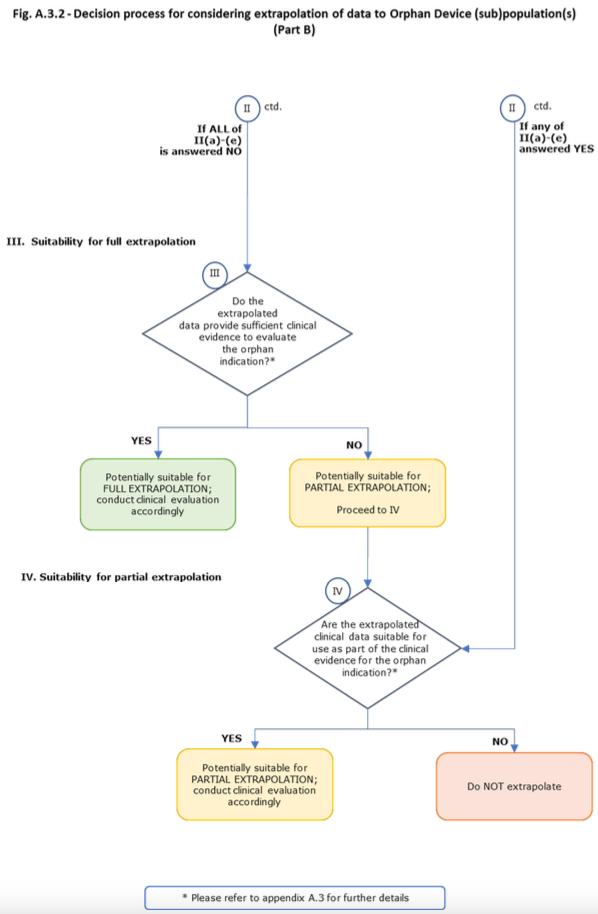
Footnotes
(1): In line with MDR Article 65 (clinical investigations on minors) and MDR Article 68 (clinical investigations in emergency situations).
(2): Article 35 of the Charter of Fundamental Rights states: “Everyone has the right of access to preventive health care and the right to benefit from medical treatment under the conditions established by national laws and practices. A high level of human health protection shall be ensured in the definition and implementation of all Union policies and activities.”
(3): MDCG 2022-14, point 18.
(4): MDR Article 2 and Annex VIII; see MDCG 2021-24
(5): Extrapolated from the population estimate criteria for Humanitarian Use Device (HUD) designation established by the U.S. Food and Drug Administration (FDA) and calculated on the basis of an EU population of 447 million, see www.fda.gov/regulatory-information/search-fda-guidance-documents/humanitarian-use-device-hud-designations
(6): Pursuant to the Council Recommendation of 8 June 2009 on an action in the field of rare diseases (OJ C 151, 3.7.2009, p. 7), for the purpose of Union-level policy work a common definition of ‘rare disease’ as a disease affecting no more than 5 per 10,000 persons should be used.
(7): For example, a cardiac valve implant may be intended to treat a rare subpopulation of patients with valvular disease which have specific anatomical characteristics, such as extreme ventricular outflow tract dilation. Other examples may include devices intended to treat a rare subpopulation of an otherwise non-rare condition, which requires definitive intervention in the neonatal population (e.g., a rare subpopulation of haemodynamically significant patent ductus arteriosus that requires acute surgical closure).
(8): Per MDR Article 5(2).
(9): I.e. for those GSPRs where clinical data are needed to demonstrate conformity.
(10): Per MDR Article 61(1).
(11): Per MDR Annex XIV.
(12): Per MDR Article 2(48).
(13): MDR Annex II, section 6.1; MDR Annex XV, Ch II, section 2.3.
(14): Including MDR Article 61(4)-(6), see MDCG 2023-7.
(15): See MDCG 2023-7.
(16): See MDR Article 56(3) and Annex VII, section 4.8.
(17): See IMDRF Principles of International System of Registries Linked to Other Data Sources and Tools (2016).
(18): Per MDR Article 83.
(19): As described in MDCG 2022-14, point 15.
(20): Per MDR Annex VII section 4.8, the notified body shall have procedures in place to allow them to decide on specific milestones for further review by the notified body of the up-to-date clinical evaluation (3rd indent) and to decide whether specific conditions or provisions need to be defined for the certification (4th indent).
(21): In accordance with MDR Article 56(3), notified bodies may require manufacturers to undertake specific PMCF studies pursuant to MDR Annex XIV, part B.
(22): See MDR Section 4.10 of Annex VII (1st subparagraph, 1st indent).
(23): See MDR Section 4.10 of Annex VII (4th subparagraph, 1st indent).
(25): The lodging of a formal application for conformity assessment is one of the conditions to benefit from the extended transitional period provided for in MDR Article 120.
(26): This possibility may be particularly useful for orphan devices for which the manufacturer has not yet submitted its clinical evaluation report to the notified body. As explained in section 8 of the Q&A on practical aspects related to the implementation of Regulation 2023/607, for the purpose of meeting the conditions for the extended MDR transitional period, the manufacturer’s application does not necessarily include the documentation that the notified body does not need for the conclusion of the written agreement and that is likely to be updated by the manufacturer before the actual conformity assessment.
(27): Commission Implementing Decision (EU) 2019/1396 of 10 September 2019 laying down the rules for the application of Regulation (EU) 2017/745 of the European Parliament and of the Council as regards the designation of expert panels in the field of medical devices (OJ L 234, 11.9.2019, p. 23).
(28): See also MDCG 2020-13, section K.
(29): Per MDR Annex XIV.
(30): This may be documented in the CER or as a separate ‘PMCF Plan’ document that is referenced in the CER.
(31): E.g., Stopping boundaries, such as O’Brien-Fleming, Pocock, or Haybittle-Peto boundaries.
(32): When appraised as required by MDR Annex XIV section 1(c).
(33): In line with MDR Annex XIV section 1(c) and 1(e).
(34): Per MDR Annex XIV part A.