
MDCG 2024-1-5 Rev.0
Guidance on the vigilance system for CE-marked devices
DSVG 05 Rev.0
Urogynaecological Surgical Mesh Implants used for Pelvic Organ Prolapse repair and Stress Urinary Incontinence
Disclaimer: This document is an interactive version of the original MDCG document. We will keep it up-to-date.
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.
Table of Contents
1. Introduction
The aim of this Device Specific Vigilance Guidance (DSVG) is to harmonise vigilance reporting and provide guidance for manufacturers of Urogynaecological Surgical Mesh Implants used for Pelvic Organ Prolapse repair and Stress Urinary Incontinence.
It provides further clarification for vigilance reporting of Urogynaecological Surgical Mesh Implants used for Pelvic Organ Prolapse repair and Stress Urinary Incontinence to the relevant Competent Authority and should be read in conjunction with the requirements of Regulation (EU) 2017/745 on medical devices (MDR) [1].
This DSVG does not replace or extend any of those requirements.
This document outlines the way to report incidents and serious incidents, defined in Article 2(64) and (65) MDR, in accordance with Articles 87 and 88 MDR, which occurred with Urogynaecological Surgical Mesh Implants used for Pelvic Organ Prolapse repair and Stress Urinary Incontinence to the relevant Competent Authority.
2. What should be reported
It is the manufacturer’s responsibility to judge each event on its own merit and to ensure compliance with the statutory reporting requirements contained within the MDR [1].
• Individual serious incident
In accordance with Article 87 MDR [1] manufacturers shall report serious incidents to the relevant Competent Authority. Serious incidents are defined in Article 2(65) MDR.
This includes circumstances where the manufacturer is uncertain whether the incident that occurred with a specific device is reportable or needs time to obtain clarification about the root cause of the incident, as indicated in Article 87(6) and (7) MDR.
The notification to the relevant Competent Authority should be reported within the timeframes referred to in Article 87(2) to (5) MDR.
For further information and clarification on what constitutes a serious incident and for details on how to apply the reporting timelines of the MDR, please refer to MDCG 2023-3 (1) “Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices” [2].
• Periodic Summary Reporting
A “Periodic Summary Report” (PSR) is an alternative reporting regime by which the manufacturer, in agreement with the respective national Competent Authority that is coordinating the periodic summary reporting (and in consultation with the Competent Authorities referred to in Article 92(8)(a) MDR), can report similar serious incidents with the same device or device type in a consolidated way.
This is possible when similar serious incidents involving the same specific device or device type occur and for which the root cause has been identified or a field safety corrective action has been implemented or where the serious incidents are common and well documented, as defined in Article 87(9) MDR.
The format, content and frequency of periodic summary reports should be agreed with the Coordinating Competent Authority (in consultation with the Competent Authorities participating in the Periodic Summary Reporting) (Article 87(9) MDR).
Until EUDAMED becomes fully functional, Competent Authorities, economic operators and other relevant parties should follow MDCG 2021-1 Rev. 1 “Guidance on harmonised administrative practices and alternative technical solutions until EUDAMED is fully functional” [3] (as required under the MDR).
• Trend Reporting
The requirements for trend reporting are outlined in Article 88 MDR [1].
In accordance with the MDR, the manufacturer should report to a Competent Authority any statistically significant increase in the frequency or severity of incidents that are not serious incidents or that are expected undesirable side-effects that could have a significant impact on the benefit-risk analysis and which have led or may lead to risks to the health or safety of patients, users or other persons that are unacceptable when weighed against the intended benefits. Trends should be identified by the manufacturer as they can be indicative of a change in the risk-benefit ratio.
For further information and clarification on what constitute incidents and undesirable side-effects please refer to MDCG 2023-3 “Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices” [2].
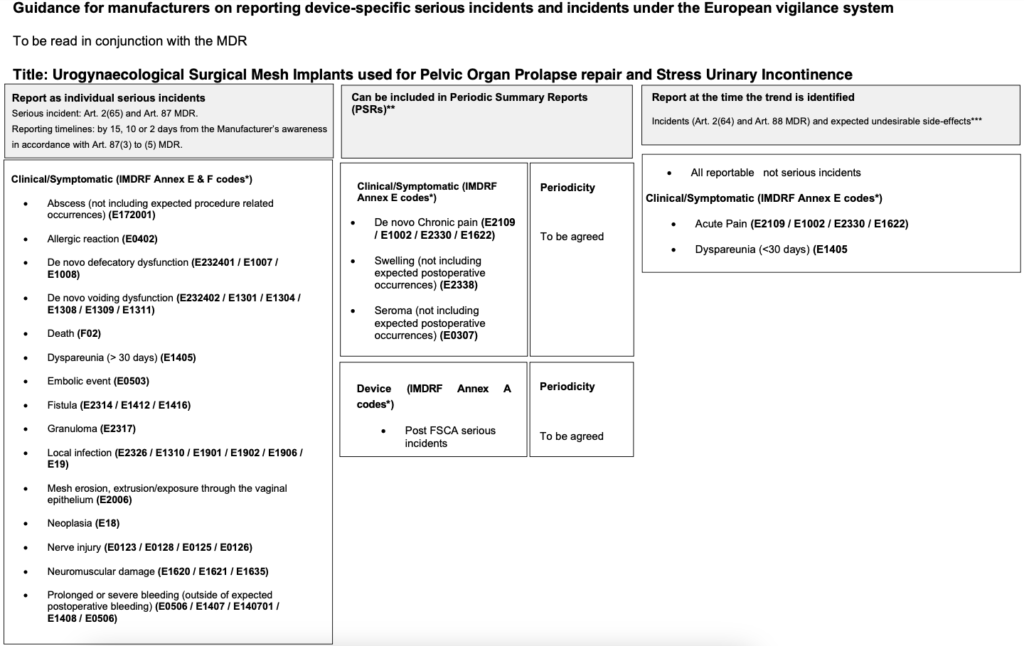
3. DSVG 05 examples
The following table details Urogynaecological Surgical Mesh Implants used for Pelvic Organ Prolapse repair and Stress Urinary Incontinence examples indicating what should be reported as device-related problems that caused or contributed to the incidents or serious incidents.
The list is for illustrative purposes only and does not constitute an exhaustive list:

4. Clinical References and Clinical Guidelines
Clinical references or current clinical guidelines for Urogynaecological Surgical Mesh Implants used for Pelvic Organ Prolapse repair and Stress Urinary Incontinence may be used by manufacturers in order to identify incident examples and complications.
Urogynaecological Surgical Mesh Implants used for Pelvic Organ Prolapse repair and Stress Urinary Incontinence manufacturers may refer to relevant local clinical guidelines when identifying incident examples and complications.
5. IMDRF Terminologies for Categorised Adverse Event Reporting
The text descriptions of Medical device problems (IMDRF Annex A) and Health effects – Clinical signs and symptoms (IMDRF Annex E) in the table are examples of what should be reported and refer to the IMDRF Annex A and E release No. 2023.
Please note that manufacturers should consult the most recent version of the IMDRF adverse event code.
The following link is provided to facilitate consultation:
https://www.imdrf.org/documents/terminologies-categorized-adverse-event-reporting-aer-terms-terminology-and-codes.
6. References
[1] Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC.
[2] MDCG 2023-3 “Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices”. Link: https://health.ec.europa.eu/system/files/2023-02/mdcg_2023-3_en_0.pdf.
[3] MDCG 2021-1 Rev. 1 “Guidance on harmonised administrative practices and alternative technical solutions until EUDAMED is fully functional”. Link: https://health.ec.europa.eu/system/files/2021-05/2021-1_guidance-administrative-practices_en_0.pdf.
Footnotes
(1): MDCG 2023-3 guidance is under revision to include IVDR aspects. Please refer to the updated version when available at the following link: https://health.ec.europa.eu/medical-devices-sector/new-regulations_en#guidance