
MDCG 2023-1 Rev.0
Guidance on the health institution exemption under Article 5(5) of Regulation (EU) 2017/745 and Regulation (EU) 2017/746
Disclaimer: This document is an interactive version of the original MDCG document. We will keep it up-to-date.
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.
Table of Contents
1 Scope and target audience
Medical devices can be manufactured and used within EU health institutions (in-house devices), on a non-industrial scale, to address the specific needs of target patient groups which cannot be met, or cannot be met at the appropriate level of performance, by an equivalent CE-marked device available on the market. In-house medical devices are exempted from most of the provisions of Regulations (EU) 2017/745 (medical devices Regulation, MDR) and (EU) 2017/746 (in vitro diagnostic medical devices Regulation, IVDR), provided the health institution adheres to the conditions laid out in Article 5(5) of the relevant Regulation. In order to ensure the highest level of health protection, Article 5(5) sets a number of rules regarding the manufacture and use of such in-house medical devices.
The provisions in Article 5(5) are the basis for the regulatory control and oversight of in-house devices. This document provides guidance on the application of some of these rules.
It is written for healthcare professionals and researchers of health institutions aiming to design, manufacture, modify and use in-house devices. In addition, this guidance document intends to foster harmonised application of Article 5(5) by the national competent authorities.
Both Regulations also state that any natural or legal person offering diagnostic or therapeutic services through distance sales to patients in the Union must use devices that comply with the MDR or IVDR (Article 6(2)). Of importance here is that the exemption provision from Article 5(5) is only applicable to health institutions within the Union.
While most recommendations in this document pertain to both medical devices and in vitro diagnostic medical devices (IVDs), some are specific to IVDs; such cases are explicitly mentioned.
Regulation (EU) 2022/112 deferred the application of some, but not all, provisions for in-house IVDs. A schematic overview of the timing of the applicability of IVDR Article 5(5) provisions can be found in Annex B of this guidance. Note that the corresponding Article 5(5) of the MDR has already been fully applicable since 26 May 2021.
2 Clarification of commonly used terms in this guidance document
- General safety and performance requirements: the relevant general safety and performance requirements of either the MDR or the IVDR are applicable to in-house devices and are laid down in Annex I of each Regulation.
- Health institution: an organisation the primary purpose of which is the care or treatment of patients or the promotion of public health (definitions 29 and 36 of the IVDR and MDR, respectively). According to recitals 29 and 30 of the IVDR and MDR, health institutions include hospitals as well as institutions, such as laboratories and public health institutes that support the health care system and/or address patient needs, but which do not treat or care for patients directly. The concept of health institution does not cover establishments primarily claiming to pursue health interests or healthy lifestyles, such as gyms, spas, wellness and fitness centres. The recognition as a health institution can also depend on national legislation and could thus differ between Member States.
- In-house device: a device that is manufactured and used only within a health institution established in the Union and that meets all conditions set in Article 5(5) of the MDR or IVDR.
- IVDR: Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU.
- MDR: Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC.
3 Guidance on terms used in Article 5(5) of the MDR and the IVDR
With the exception of the relevant general safety and performance requirements set out in Annex I, the requirements of the MDR and IVDR shall not apply to devices manufactured and used only within health institutions established in the Union, provided that specific conditions are met.
3.1 Which devices are referred to in Article 5(5)?
3.1.1 MDR
According to Article 1(4) of the MDR, the term ‘devices’ means (1) medical devices, (2) accessories for medical devices and (3) products listed in Annex XVI.
(1) ‘medical device’ is defined in Article 2(1) of the MDR.
(2) ‘accessory for a medical device’ is defined in Article 2(2) of the MDR.
(3) ‘products listed in Annex XVI’ refers to the groups of products without an intended medical purpose that are listed in Annex XVI of the MDR. Applicability of the MDR to these products, and therefore application of Article 5(5), will be effective from the date of application of common specifications for these products.
Note:
Custom-made devices are out of scope of Article 5(5) and should follow the relevant requirements of the MDR.
3.1.2 IVDR
According to Article 1(2) of the IVDR: the term ‘devices’ means (1) IVDs and (2) accessories for IVDs.
(1) ‘IVD’ is defined in Article 2(2) of the IVDR.
(2) ‘accessory for an IVD’ is defined in Article 2(4) of the IVDR.
3.1.3 General
Whenever the term ‘device’ is used in this guidance document, it refers to the definitions in Article 1(4) and Article 1(2) of the MDR and IVDR, respectively.
Note:
- A protocol, in the form of a written procedure (in paper or digital format, e.g. describing the manufacture or use of an in-house device), that is shared between health institutions is not considered as a device according to the definitions above. Consequently, the MDR and IVDR do not apply to such protocols.
- Patient specimens are not considered as devices according to the definitions above. Consequently, the MDR and IVDR do not apply to these and patient specimens can be shared between health institutions.
- While delivery of the results (reading the results from the in-house device) should occur in the health institution that manufactured the in-house device, this should not preclude the sharing of results between health institutions or with the patient.
- Any product or a combination of products that meets the definition of ‘device’ must comply with the MDR or IVDR; i.e. either be CE marked, or be manufactured in-house by a health institution and thus comply with Article 5(5), or be an investigational device (MDR) or a device for performance study (IVDR), or be a custom-made device (MDR), or be exceptionally allowed a derogation from CE marking by a competent authority.
- According to the second paragraph of Article 5(5), member states retain the right to restrict the manufacture and the use of any specific type of in-house devices. Health institutions are advised to consult national legislation or may contact their competent authority for possible restrictions in their country.
3.2 How to understand the terms ‘manufactured and used’?
A device must be manufactured and used only within the same health institution in order for Article 5(5) to apply.
3.2.1 How to understand the term ‘manufactured’?
Manufacturing a device by a health institution can include:
- manufacturing a device from raw materials, from parts or components of a device or of another type of product or from an existing device or another type of product,
- combining a device with another device or another type of product, where the combination creates a new device,
- modifying an existing device in order to create a new device.
3.2.2 How to understand the term ‘used’?
Devices can only be defined as in-house devices when their manufacture and use is limited to health institutions established in the Union. This use within health institutions can either be physical or, for instance for medical device software, remote, provided they are not made available to another legal entity (see section 3.4 in this guidance). The act of using an in-house manufactured device is performed within the health institution when the device is used in the care or diagnosis of a patient. If, during the lifecycle of the device, the device is used outside the health institution’s legal entity, it cannot be in-house.
Notes:
- The IVDR and MDR generally do not regulate the actual use of a device by a healthcare professional. Nevertheless, the use of CE marked devices by healthcare professionals outside the manufacturer’s intended purpose may be subject to national provisions.
However, where a health institution (as opposed to an individual healthcare professional) changes the intended purpose of a CE-marked device, in view of its use in the health institution with that changed intended purpose, then Article 5(5) applies. - RUO (research use only) products are not regulated by the MDR and IVDR and are not considered to be in-house devices when they are used for research purposes only.
However, where a health institution (as opposed to an individual healthcare professional) ascribes to a RUO product an intended purpose such that it would meet the definition in Article 2(1) MDR or 2(2) IVDR in view of its use in the health institution, then Article 5(5) applies. National legislation on quality of care may also apply.
In‐house devices may include RUO products as components, provided that the resulting in‐house device complies with the requirements of Article 5(5).
3.2.3 General
Examples of devices that would fall under the definition of an in-house device:
- PCR master mix: a health institution orders primers based on scientific literature and manufactures its own in-house master mix containing buffer, primers, dNTPs, cofactors and enzymes to run PCRs on human DNA/RNA specimens.
- A health institution develops in-house a medical device software that is used on site by healthcare professionals.
Examples of devices that do not fall under in-house devices:
- Medical device applications where patients can use the application outside the health institution, e.g. by entering or accessing medical data that are subsequently used by the healthcare professional.
- Orthopaedic braces that can be adapted (and thus ‘used’) by patients themselves outside the health institution.
- Self-tests cannot fall under Article 5(5) if used outside the health institution’s legal entity. However, an in-house manufactured self-test can be used within the health institution by lay persons. Also, an in-house device can be used in the health institution’s laboratory for the analysis of a specimen that is collected by patients themselves and consecutively sent to the laboratory.
- Devices manufactured in-house purely for economic motives/financial interests, without clinically relevant reasons.
3.3 Compliance with the relevant general safety and performance requirements
Health institutions must ensure that their in-house manufactured devices are compliant with the relevant general safety and performance requirements in Annex I of the MDR and IVDR. Some important aspects of this are described below.
- Chapter I of Annex I describes the establishment of a risk management system and the regular update of the benefit-risk ratio assessment. Note that device risks do not only pertain to patient risks, but also to risks to the users, as well as to risks related to use error.
- Chapter II of Annex I describes requirements regarding design, manufacture and performance of devices and is therefore also particularly relevant to in-house devices. Health institutions should carefully check what provisions apply to their in-house devices as this information will also be paramount to establishing proof on non- equivalency of available CE marked devices (see section 3.6 in this guidance).
- Chapter III of Annex I defines requirements for the information that is supplied with the device. While a number of the provisions in this chapter do not apply to in-house devices, some are relevant to safely use the device in a way that allows it to achieve its intended purpose, e.g.:
– operating instructions/protocols,
– information on substances or mixtures which may be considered as being dangerous,
– expiry or production dates of the manufactured devices or batches,
– storage and handling conditions,
– the lot/serial number or an equivalent means of identification for traceability purposes.
Note:
It is essential that health institutions properly document and regularly update the proof of compliance of their in-house devices with Annex I as this documentation contains critical information that will be used by competent authorities to assess compliance with Article 5(5). Additionally, critical changes made to in-house devices should be evaluated and documented.
3.4 Legal entity
Reference to the IVDR/MDR: Article 5(5)(a).
In-house devices shall not be transferred to another legal entity.
Healthcare systems are organised differently in different member states. Therefore, the concept of legal entity can differ. The national competent authority may clarify how legal entity is understood nationally.
Examples of legal entities:
- One hospital (1) can be one legal entity when there is only one health institution (one organiser) within the hospital.
- One hospital (1) can accommodate several legal entities when there are different health institutions (different organisers) within the same hospital. The different health institutions can have different organisational numbers and different quality management systems.
- Several hospitals (1) can belong to the same legal entity when they are all part of one health institution (one organiser). They share the same organisational number, quality goals and quality management systems and the same healthcare strategy, even though they might be spread over different locations.
3.5 What is an appropriate quality management system?
Reference to the IVDR/MDR: Article 5(5)(b).
The manufacture and use of in-house devices must occur under an appropriate quality management system (QMS).
3.5.1 MDR
The QMS should be in compliance with Article 5(5), the relevant requirements of Annex I, national legislation and applicable ISO-standard(s) if the health institution is certified/accredited.
Article 10(9) of the MDR describes the minimal aspects that a QMS for manufacturing CE-marked medical devices should cover. This Article can be used as guidance (with some MDR-specific exceptions) on what an appropriate QMS for manufacturing in-house devices is. If applicable, ISO-standards concerning for instance manufacturing and risk management can be used, especially if harmonized with the MDR.
Examples of areas covered by an appropriate QMS:
- Compliance to Article 5(5) and Annex I, MDR
Health institutions must determine how the requirements of Article 5(5) will be met.
A declaration must be drawn up and published (e.g. via the health institution’s web site, see also section 3.8 of this guidance), confirming compliance with the general safety and performance requirements of Annex I, Article 5(5) (e). To make this declaration, there is a need to identify the applicable general safety and performance requirements and to document compliance with these relevant requirements for the device in question (see also section 3.3 of this guidance).
- Responsibility of the management
This includes resource management.
According to Article 5(5) (g), the health institution must take all necessary measures to ensure that all devices are manufactured in accordance with the documentation referred to in Article 5(5) (f).
- Risk management
Health institutions that manufacture in-house devices must establish, implement, document and maintain a risk management system. Risk management must be understood as a continuous iterative process throughout the entire lifecycle of a device, requiring regular systematic updating, as per Point 3 Annex I.
- Identify, generate and appraise data
There is a need for supporting data and analysis of that data to justify that the target patient group´s specific needs cannot be met or cannot be met at the appropriate level of performance in another way than by manufacturing and using the health institution´s device. The experience gained from clinical use of the device should be used to review the device performance, Article 5(5) (c), (f) and (h).
- Manufacturing
The health institution must draw up documentation that makes it possible to have an understanding of the manufacturing facility, the manufacturing process, the design and performance data of the devices, including intended purpose, Article 5(5) (f).
- Traceability
According to Article 5(5) (e) (ii), health institutions must make publicly available the details necessary to identify the devices. Chapter III, Annex I describes labelling. The system of identification of devices links to Article 5(5) (h), which states that health institutions shall take all necessary corrective actions. So, the identification of the device can be a part of a product tracking system to identify the affected products and involved patients.
- Monitoring, analysis and continuous improvement
The health institution must review experience gained from clinical use of the device and take all necessary corrective actions, Article 5(5) (h).
- Communication with competent authorities
According to Article 5(5) (d), (f) and the second last subparagraph of 5(5), the health institution must be prepared to provide detailed information to the competent authority.
3.5.2 IVDR
The laboratory of the health institution must be compliant with standard EN ISO 15189 or where applicable national provisions, including national provisions regarding accreditation and certification.
Article 10(8) of the IVDR describes the minimal aspects that a QMS for manufacturing CE-marked medical devices should cover. This Article can be used as guidance (with some IVDR-specific exceptions) on what an appropriate QMS for manufacturing in-house devices is. If applicable, ISO-standards concerning for instance manufacturing and risk management can be used, especially if harmonised with IVDR.
Examples of areas covered by an appropriate QMS:
- Compliance to Article 5(5) and Annex I, IVDR
Health institutions must determine how the requirements of Article 5(5) will be met.
A declaration must be drawn up and published (e.g. via the health institution’s web site, see also section 3.8 of this guidance), confirming compliance with the general safety and performance requirements of Annex I, Article 5(5) (f). To make this declaration, there is a need to identify the applicable general safety and performance requirements and to document compliance with these relevant requirements for the device in question (see also section 3.3 of this guidance).
- Responsibility of the management
This includes resource management.
According to Article 5(5) (h), the health institution must take all necessary measures to ensure that all devices are manufactured in accordance with the documentation referred to in Article 5(5) (g).
- Risk management
Health institutions that manufacture in-house devices must establish, implement, document and maintain a risk management system. Risk management must be understood as a continuous iterative process throughout the entire lifecycle of a device, requiring regular systematic updating, as per Point 3 Annex I.
- Identify, generate and appraise data
There is a need for supporting data and analysis of that data to justify that the target patient group´s specific needs cannot be met or cannot be met at the appropriate level of performance in another way than by manufacturing and using the health institution´s device. The experience gained from clinical use of the device should be used to review the device performance, Article 5(5) (d), (g*) and (i).
* applies only to class D devices unless regulated otherwise by national provisions.
- Manufacturing
The health institution must draw up documentation that makes it possible to have an understanding of the manufacturing facility, the manufacturing process, the design and performance data of the devices, including intended purpose, Article 5(5) (g*).
* applies only to class D devices unless regulated otherwise by national provisions.
- Traceability
According to Article 5(5) (f) (ii), health institutions must make publicly available the details necessary to identify the devices. Chapter III, Annex I describes labelling. The system of identification of devices links to Article 5(5) (i), which states that health institutions shall take all necessary corrective actions. So, the identification of the device can be a part of a product tracking system to identify the affected products and involved patients.
- Monitoring, analysis and continuous improvement
The health institution shall review experience gained from clinical use of the device and take all necessary corrective actions, Article 5(5) (i).
- Communication with competent authorities
According to Article 5(5) (e), (g*) and the second last subparagraph of 5(5), the health institution must be prepared to provide detailed information to the competent authority.
* applies only to class D devices unless regulated otherwise by national provisions.
Note:
For in-house IVDs, the laboratory of the health institution should be in compliance with the standard EN ISO 15189 or with national provisions, including national provisions regarding accreditation. Compliance with the standard EN ISO 15189 may be understood as accreditation to the standard or other means of compliance. However, as the manufacturing process of a device and the compliance to the relevant requirements of Annex I is not in the scope of this standard, compliance with EN ISO 15189 alone does not constitute an appropriate QMS for the manufacture of in-house IVDs.
3.5.3 General
For both medical devices and IVDs, the QMS can cover the whole health institution or parts of the health institution involved in the manufacturing, modification and use of the in-house device. A QMS should include a process for obtaining information about equivalent CE- marked devices that become available on the market (see section 3.6 in this guidance).
3.6 Justification that the target patient group’s specific needs cannot be met, or cannot be met at the appropriate level of performance, by an equivalent device available on the market
Reference to the IVDR/MDR: Article 5(5)(d)/(c).
The health institution must justify in its documentation that the target patient group’s specific needs cannot be met, or cannot be met at the appropriate level of performance by an equivalent device available on the market.
3.6.1 Target patient group’s specific needs
In this context, the target patient group should be understood as a group of patients who have in common the same disease, condition or characteristics, and who could benefit from using the device.
The specific needs should be understood as needs for:
- a specific device (as clarified in section 3.1 of this guidance) or;
- a specified level of performance of a device for certain performance characteristics.
Examples:
- The health institution’s target population is paediatric while CE marked devices are only targeted at the adult population and there are significant differences in the values between adults and children (e.g. some hormone levels).
- Oestradiol can be measured in healthy females with commercial immunoassays, however in breast cancer females on aromatase inhibitors, a more sensitive LCMS method is needed.
- For a particular patient group, an in-house IVD usefully combines the analysis of 2 or more CE marked IVDs, thereby avoiding excess, sometimes painful, patient specimen taking.
3.6.2 Equivalent device in the context of the justification
3.6.2.1 MDR
Annex XIV.3 of the MDR describes device characteristics that should be taken into consideration for the demonstration of (non-)equivalence. These characteristics are divided into technical, biological and clinical aspects.
- Technical: the device is of similar design, is used under similar conditions, has similar specifications and properties including physicochemical properties, uses similar deployment methods, has similar principles of operation and critical performance characteristics.
- Biological: the device uses the same materials or substances in contact with the same human tissues or body fluids for a similar kind and duration of contact, has similar release characteristics of substances, including degradation products and leachables.
- Clinical: the device is used for the same clinical condition or purpose, including similar severity and stage of disease, at the same site in the body, in a similar population, has the same kind of user, has a similar relevant critical performance in view of the expected clinical effect for a specific intended purpose.
Note:
There is a different usage of the terms ‘similar’ and ‘same’ in the device characteristics described above. The health institution should consult the ‘MDCG 2020-5 guidance on Clinical Evaluation – Equivalence’ for further guidance on the subject. A justification of non-equivalence based on technical, biological or clinical aspects of the device should be documented.
3.6.2.2 IVDR
The IVDR does not provide a description of equivalent devices. However, some of the equivalence characteristics listed in the MDR are also applicable to IVDs (see above). Justification that the patient group’s specific needs cannot be met, or cannot be met at the appropriate level of performance, by an equivalent device available on the market can be based on technical, biological or clinical aspects e.g. different intended purpose, different clinical conditions, different patient group, different conditions of use, different principles of operation, different approved specimen materials, different critical performance characteristics or different critical technical specifications.
A justification of non-equivalence based on technical, biological or clinical aspects of the device should be documented.
3.6.3 Process for producing and reviewing the justification
Before manufacturing an in-house device for the first time, a health institution should examine the market for the presence and availability of equivalent CE-marked devices. It is appropriate to describe this process in the QMS for the in-house device. The European database on medical devices, EUDAMED, could serve as one of the sources of information for the identification of equivalent CE-marked alternatives (e.g. for higher risk class devices, a summary of safety and (clinical) performance is publicly available in EUDAMED). Other sources of information comprise information from manufacturers, distributors, scientific conferences etc. On the basis of its findings, the health institution should justify why the target patient group’s specific needs cannot be met, or cannot be met at the appropriate level of performance, by an equivalent device available on the market. This justification should be documented.
The health institution should continue gathering information about the availability on the market and performance of potentially equivalent CE-marked devices in order to keep their in-house device manufacturing up-to-date with market developments. The health institution should review its justification on a regular basis, in view of its findings.
Once the in-house device is in use, a possible subsequent availability on the market of an equivalent device does not invalidate the initial justification regarding the fulfilment of the requirements set out in Article 5(5) at moment of the start of the in-house manufacturing. However, in such a case the health institution should review and update its justification. If a CE-marked device turns out to be at least equivalent to the in-house device and meets the target patient group’s specific needs at the appropriate level of performance, a transition process towards the usage of the CE marked device should start.
3.6.4 Availability on the market
Market in this context should be understood as the market of CE-marked devices in the relevant member state. Availability means that the equivalent device is accessible to the health institution according to EU, national and local rules and regulations. EUDAMED will be one of the main sources of information regarding availability on the market.
3.7 What kind of information can be requested from health institutions by competent authorities?
Reference to the IVDR/MDR: Article 5(5)(e)/(d).
The health institution must provide information upon request on the manufacture and use of in-house devices to its competent authority, which must include a justification of their manufacturing, modification and use.
Examples of what information can be requested:
- When an in-house device is put into service:
– device type,
– intended purpose,
– target patient group,
– data on the design, on safety and performance and on the expected benefit from the device,
– a justification for the lack of equivalent CE-marked alternatives, or the lack of equivalent CE-marked alternatives with an appropriate level of performance, that meet the target patient group’s specific needs,
– description of the manufacturing process,
– description of modifications performed,
– information regarding use: procedures, use in combination with other devices (data on compatibility) etc.
- After the device has been used routinely:
– all information as described above,
– number of units or batches manufactured in a certain period and a justification of the production numbers,
– data regarding the performance of the device during routine use: performance outcome, incidents or complaints, corrective actions undertaken.
Note:
Health Institutions should check whether national provisions exist on notifying the competent authority when an in-house device is put into service, modified or its use discontinued.
3.8 Public declaration
Reference to the IVDR/MDR: Article 5(5)(f)/(e).
The health institution must draw up a declaration which it must make publicly available, including:
- the name and address of the manufacturing health institution,
- the details necessary to identify the devices (product name, product code or reference, description or other unambiguous reference allowing the identification of the device, intended use),
- a declaration that the devices meet the general safety and performance requirements set out in Annex I of the IVDR or MDR and, where applicable, information on which requirements are not fully met with a reasoned justification therefor.
Health institutions should consult possible national legislation, rules or guidance regarding the exact declaration format, language requirements and the publication conditions that need to be fulfilled (e.g. publication on the health institution’s website and/or on a dedicated webpage from the competent authority). A proposed declaration format is provided in Annex A of this guidance. Health institutions should regularly review their public declaration and update it as necessary.
3.9 Documentation requirements
Reference to the IVDR/MDR: Article 5(5)(g)/(f).
For all medical devices and for class D IVDs (or any other IVD class if obliged by national legislation), the health institution must draw up documentation that makes it possible to have an understanding of the manufacturing facility, the manufacturing process, the design and performance data of the devices, including the intended purpose, and that is sufficiently detailed to enable the competent authority to ascertain that the general safety and performance requirements set out in Annex I of the MDR and IVDR are met. Health institutions should consult national provisions regarding possible documentation requirements for class A, B and C IVDs.
The following aspects (non-exhaustive list of examples that might be applicable) should be taken into account when drafting documentation for in-house devices.
- Manufacturing facility: description of the infrastructure, the services and the work environment needed to safely manufacture the device in a way that the product fulfils the requirements, listing of the equipment that is essential for production etc.
- Manufacturing process: explanation of the manufacturing processes, including a description of the raw materials, control of suppliers, final product testing etc.
- Design: principles of operation of the device and its mode of action, technical specifications including chemical, physical and biological properties, technology used, software development, listing of applied standards, common specifications and guidelines that are essential to meet the relevant general safety and performance requirements etc.
- Performance data: according to Annex I of the IVDR/MDR, devices shall be designed and manufactured in such a way that they are suitable with regard to the performance they are intended to achieve, taking account of the generally acknowledged state of the art. A description of, where applicable, the analytical and the clinical performance data supporting the intended purpose should be provided.
- Intended purpose of the device: specification of indications and contra-indications, the patient target group or groups, information provided by the device, function of the device (e.g. screening, monitoring, diagnosis, …), what type of specimen is used by an IVD device etc.
All information should be presented in a clear, organised, readily searchable and unequivocal way. Annex II of the MDR/IVDR can be used as guidance for documentation purposes. The documentation must be kept up to date.
3.10 Vigilance, incidents and corrective actions
Reference to the IVDR/MDR: Article 5(5)(i)/(h).
The health institution must review experience gained from clinical use of the devices and take all necessary corrective actions.
Health institutions should have a documented procedure in place to collect clinical and performance data and to process incidents and corrective actions for in-house devices. They should consult national legislation on possible requirements regarding reporting of incidents and corrective actions.
3.11 Industrial scale
The last sentence of Article 5(5) of both Regulations states that the exemption provisions do not apply to devices manufactured on an industrial scale. Furthermore, the recitals of the Regulations state that healthcare institutions should be able to manufacture, modify and use devices in-house and thus meet, on a non-industrial scale, the specific needs of a patient group that cannot be met at an appropriate level of performance by an equivalent device available on the market.
There is no definition of the term ‘industrial scale’ in the MDR or the IVDR. It should be distinguished from the term ‘mass-produced’. The concept is a combination of many factors to be considered on a case-by-case basis, including e.g. volume of production, commercial aspects and manufacturing process.
The exemption provisions in Article 5(5) of the Regulations should only be applicable to devices that are produced by the health institutions in order to meet the patient groups’ specific needs, and therefore, the manufacturing process should not produce more than the estimated number of required devices.
Note:
- ‘Industrial scale’ is not synonymous to ‘mass-produced’. A mass-produced medical device is defined in the IMDRF/PMD WG/N49:2018 document as a medical device that is based on standardised dimensions/designs; that is not designed for a particular individual; and that is typically produced in a continuous production run or homogenous batch.
- In case of IVDs, the analysis of a large number of patient specimens does not automatically render an in-house IVD to a device produced on an industrial scale.
Annex A
Public declaration regarding the manufacture and use of in-house devices by health institutions
Name of health institution:
Address:
-the health institution- declares that the devices described in the accompanying table are only manufactured and used in -the health institution- and do meet the applicable general safety and performance requirements (GSPR) of the medical devices Regulation (EU 2017/745) or of the in vitro diagnostic medical devices Regulation (EU 2017/746). A reasoned justification is provided in case applicable general safety and performance requirements are not fully met.
Date and location:
Name, function and signature of responsible person(s):
Table of in-house devices:
| Device identification (e.g. name, description, reference number) | Device type (IVD/MD) | Risk class of the device (2) | Intended purpose | Applicable GSPR fully met? (Y/N) | Information on and justification for applicable GSPR that are not fully met (using the numbering as in Annex I of the IVDR/MDR) |
|---|---|---|---|---|---|
Annex B
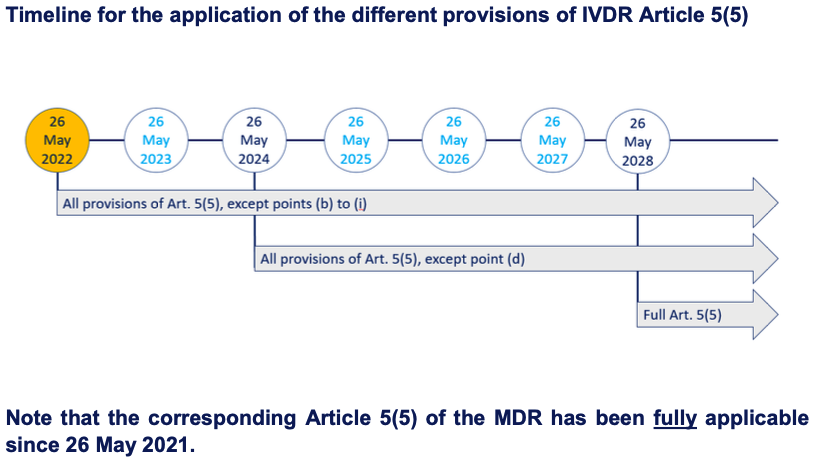
Footnotes
(1): The term ‘hospital’ can be exchanged for any of the examples provided in the definition of health institution as described in section 2.
(2): The risk class of the device can be determined by using Annex VIII of the IVDR and MDR. MDCG 2020-16 and MDCG 2021-24 provide further guidance on classification of IVDs and medical devices, respectively.
Revision History
January 2023