
MDCG 2022-21 Rev.0
GUIDANCE ON PERIODIC SAFETY UPDATE REPORT (PSUR) ACCORDING TO REGULATION (EU) 2017/745 (MDR)
Disclaimer: This document is an interactive version of the original MDCG document. We will keep it up-to-date.
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.
Table of Contents
1 Introduction
1.1 What is the Periodic Safety Update Report
The Periodic Safety Update Report (PSUR) has been introduced in Article 86 of the Medical Device Regulation (MDR) 2017/745. The introduction of the PSUR requirement under the MDR requires a more consistent, standardized and systematic review of all Post-Market Surveillance (1) (PMS) data by manufacturers of class IIa, class IIb and class III devices.
The PSUR summarises the results and conclusions of the analysis of the post-market surveillance data gathered as a result of the Post-Market Surveillance Plan (see section 2.1).
The range of PMS data and level of detail of information examined in a PSUR, the modalities for aggregating and assessing the data or the grouping of devices within the same PSUR will depend mainly on the type of device(s) covered, the time span during which the manufacturer has placed them on the EU market and the post-market experience gained with these devices.
The main objective of this guidance document is to assist manufacturers to implement the legal requirements laid down in Article 86 MDR. However, manufacturers should have reasonable time to adapt their quality management systems and sufficient flexibility (2) when they draw up and update a PSUR as long as they can demonstrate that it is in line with Article 86 MDR.
Moreover, PSURs already drawn up or in the process of being prepared when this guidance is published should not be expected to have followed this guidance, as long as those PSURs comply with the legal requirements laid down in Article 86 MDR.
1.2 Terminology
“Device certification date”: The “device certification date” used in this guidance should be understood as one of the following:
– Date of issuance of the EU technical documentation assessment certificate according to Annex IX (including chapter II).
– Date of issuance of the EU type-examination certificate according to Annex X.
– Date of issuance of the EU product verification certificate for class IIa devices according to section 3 of Annex XI.
– The date of signing of the Declaration of Conformity for Annex IX (Chapters I & III) devices.
– Date of the issuance of the statement required in Annex XIII, section 1 for custom- made devices (3).
Legacy devices (4) should be understood as devices (5), which, in accordance with Article 120(3) of the MDR, are placed on the market after the MDR date of application (DoA) and until the end of the transition period laid down in Article 120(3) MDR if certain conditions are fulfilled.
Those devices can be:
-> devices which are class I devices under Directive 93/42/EEC (MDD), for which an EC declaration of conformity was drawn up prior to 26 May 2021 and for which the conformity assessment procedure under the MDR requires the involvement of a notified body;
-> devices covered by a valid EC certificate issued in accordance with Directive 90/385/EEC (AIMDD) or the MDD prior to 26 May 2021.
Leading device: The “leading device” in a group of devices covered by the same PSUR corresponds to the highest risk class device. In the case when there are several devices with the same risk classification, the manufacturer should assign a leading device.
Old devices (4) are those devices that were placed on the market before 26 May 2021 in accordance with the AIMDD or the MDD or in accordance with the applicable rules before the Directives had entered into force.
PSUR Web Form: Template that contains all the information that will be available in Eudamed for the PSUR. This form details information regarding the medical device, manufacturer, NB and the management of the PSUR process.
PSUR reference number: The PSUR reference number is the unique identifier that the manufacturer must assign to a PSUR. It should remain the same during the whole PSUR lifetime.
PSUR version number: an incremental number attributed to each update(s) of the PSUR which has been made available by a manufacturer and allowing to identify and trace them.
The term “annually” for drawing up the first PSUR for legacy devices should be understood as occurring during the course of the calendar year following MDR date of application.
The term “every two years” for drawing up the first PSUR for legacy devices should be understood as occurring during the course of the second calendar year following MDR date of application.
1.3 Scope of the guidance
This guidance is applicable to medical devices which have been certified under the MDR and to devices which have been certified under MDD 93/42/EEC or AIMDD 90/385/EEC except for devices which have stopped being placed on the EU market before the MDR date of entry into application (DoA) (see section 3.1). It does not apply to in vitro diagnostic medical devices which have been certified under IVDR (EU) 2017/746 or IVDD 98/79/EEC for which a specific guidance is intended.
Manufacturers of class I devices do not have to prepare a PSUR; instead, they should prepare a Post-Market Surveillance Report (PMSR) as detailed in Article 85. This guidance, although not covering PMSR, may provide useful suggestions on how information can be presented.
2 General considerations
2.1 PSUR objectives
The PSUR objectives are double:
2.1.1 Identification and evaluation of changes of the benefit-risk profile
The main objective of a PSUR is to present a summary of the results and conclusions of the analyses of post-market surveillance data relating to a device or a device group, thus allowing the reporting of any possible changes to the benefit-risk profile of the medical device(s), considering new or emerging information in the context of cumulative information on benefits and risks.
Manufacturers should present this information to identify any safety and performance concerns, through both reactive and proactive PMS data collection. When concerns have been identified this gathered information should be used to re-evaluate the benefit-risk profile and the state of the art of the medical device(s).
When there is evidence of an adverse change to the benefit-risk profile of the medical device(s), this information should be evaluated and considered in line with the clinical evaluation and Risk Management. In the event of such circumstances, there should be clear consideration and evaluation as to whether the medical device remains safe and effective (see also section 2.2.2).
In line with the objectives of the MDR, the PSUR should provide transparency of all Post Market Surveillance data to the Notified Body involved in the conformity assessment of the device and to the Competent Authorities.
The PSUR should summarize the results and conclusions of the analysis of the data that the manufacturer has systematically and actively gathered in post-market surveillance with its device(s) and, where relevant, with similar devices.
2.1.2 Information on Preventive or Corrective Actions (CAPA) according to Article 83(4)
The PSUR may also be the tool to provide information about Corrective Action(s) or Preventive Action(s) (CAPA) which are covered by Article 83(4) first sentence and for which the information of the competent authorities and, when applicable, the notified body about these CAPAs and their implementation is required (6). The actions referred to in Article 83(4) second sentence i.e. serious incidents and field safety corrective actions, are intended to be reported through EUDAMED.
The scope of the various types of CAPAs under Article 83(4) does not cover quality management system related CAPA’s unless these could have a direct impact on product safety, performance or quality.
These CAPA can be related to:
- Devices already placed on the EU market.
- Issues that might have a direct impact on the product and that might impact product safety, performance or quality and,
- Any action related to a voluntary and non-temporary suspension of marketing of the device by the manufacturer which is not related to a commercial decision.
- Evaluation of benefits and risks identified through post-market activities as described in Annex III, point 1 (a) of MDR, i.e. in particular:
- records referring to non-serious incidents and data on any undesirable side- effects;
- relevant specialist or technical literature, databases and/or registers;
- information, including feedbacks and complaints, provided by users, distributors and importers and,
- publicly available information about similar medical devices.
A summary of all the above Article 83(4) first sentence CAPA can be made available to the Competent Authorities either through the PSUR or through a specific report. However, all safety related CAPA should be part of the PSUR (see section 2.2).
2.2 PSUR content
2.2.1 General aspects
For the purpose of the PSUR guidance, the term “device” relates to a device model and not to an “individual” device, as “individual” devices are placed on the market at different moments during the period covered by the “device” certificate.
Per Article 86(1) and Annex III, point 2 of the MDR, the PSUR is part of the technical documentation on post-market surveillance to be drawn up by the manufacturer in accordance with Articles 83 to 86. The PSUR should be presented in a clear, organized, readily searchable and unambiguous manner.
The PSUR should be generated as a stand-alone document that can be assessed independently from the supporting documentation. The PSUR should provide a general overview of all post-market surveillance activities and the data collected and analysed based on the PMS plan for the device. Therefore, the aim of the PSUR is not to duplicate all data and reports generated by the PMS Plan but to summarize all results and conclusions.
The manufacturer should specify the relevant information and sections of the different reports and provide a summary of the data collected, their assessment and conclusion as well as the actions taken when appropriate. If a manufacturer decides that specific datasets are not used or deemed not to be required, the manufacturer should duly justify why these datasets are not included in the PSUR.
It is recommended to add an executive summary in particular as regards the main relevant information related to benefits and risks and to the changes in the acceptability of the benefit-risk profile.
To the extent possible, a similar presentation of the PSUR should be followed regardless of the device class. A recommended template for the PSUR is provided in Annex I of this guidance.
2.2.2 Specific aspects
In accordance with Article 86(1) MDR, the PSUR should summarize the results and conclusions of the analysis of the post-market surveillance data gathered as a result of the PMS Plan and provide:
- the conclusions of the benefit-risk determination (7);
- the main findings of the Post-Market Clinical Follow-up (PMCF);
- the volume of sales of the device and an estimate evaluation of the size and other characteristics of the population using the device and, when practicable, the usage frequency of the device.
In order to prepare the above summary, the following elements should be considered:
- Information concerning serious incidents and field safety corrective actions;
- records referring to non-serious incidents and data on any undesirable side- effects;
- information from trend reporting;
- relevant specialist or technical literature, databases and/or registers;
- information, including feedbacks and complaints, provided by users, distributors and importers;
- publicly available information about similar medical devices.
More detailed information on (i) the collection of the relevant data and (ii) their presentation and evaluation are provided respectively in Annex I and Annex III of this guidance.
3 Scope and duration of the PSUR requirement
3.1 Scope of the PSUR requirement
3.1.1 Devices within the scope of PSUR requirement
Article 86 requires manufacturers of class III, class IIb and class IIa devices to “prepare a Periodic Safety Update Report (PSUR) for each device and when relevant, for each category or group of devices”.
- MDR compliant devices
- Class IIa, class IIb and class III devices certified according to the requirements of the MDR placed on the market or put into service either before or after the MDR Date of Application, 26 May 2021 (DoA);
- Custom-made devices falling within class IIa, class IIb and class III complying with the requirements of the MDR;
- Annex XVI devices falling within class IIa, IIb and class III devices, once the MDR becomes applicable to those devices;
- “Legacy devices”
Taking into account that the term “device” in this guidance relates to a device model and not to an “individual” device (see section 2.2.1.) and that “individual” devices may have been placed on the market both before and after MDR DoA based on Article 120(3) regime, the scope of “legacy devices” covers all the devices for which at least some individual devices have been placed on the market before MDR DoA.
3.1.2 Devices outside the scope of PSUR requirement
- Class I MDR devices and class I legacy devices.
- “Old devices”: the scope of “old devices” covers only the devices for which no individual devices have been placed on the market from MDR DoA.
3.2 Duration of the PSUR requirement
3.2.1 Device lifetime
A PSUR is required throughout the lifetime of the device: The lifetime of a device is the time period specified by the manufacturer in the device documentation during which the device is expected to remain safe and effective for use / in use. If the lifetime exceeds the administrative provisions in Annexes IX, X, XI, XIII of MDR, i.e. max 10 years for non-implantable devices and max 15 years for implantable devices, then the shorter timeline between the lifetime of the device and the retention period should prevail.
3.2.2 End of obligation to update the PSUR
Determination of the end of the PSUR requirement:
A PSUR is no longer required to be updated when the last manufactured device of the device model has been placed on the market and the intended lifetime of that (individual) device; i.e. the overall lifetime of the device (model), has been achieved. The overall lifetime of a device (model) should include the time between the placing on the market of the last manufactured device and the end of the intended lifetime of that (individual) device.
Example 1: A single-use device may have an intended lifetime shorter than its shelf-life. After the placing on the market of the last device of the final production cycle, the PSUR can be terminated when the end of the shelf-life plus intended lifetime has been reached.
Example 2: An X-ray machine may have no shelf-life but should have a lifetime determined by its continuation to perform and remain safe as intended. A PSUR is required until the end of the intended lifetime of the last device placed on the market.
Example 3: An implantable device may have a shelf-life of 3 years but an intended lifetime of 10 years. After the last device produced has been placed on the market, the PSUR can be terminated when the shelf-life (3 years) plus the intended lifetime (10 years), i.e. 13 years, has passed.
When a device’s certificate has expired and the lifetime of the device has not yet been covered by the last PSUR, a PSUR should continue to be made available, upon request, to the competent authorities.
When the placing of the device on the market has been discontinued by the manufacturer while the device’s certificate has not yet expired, a PSUR should continue to be made available to the notified body involved in the conformity assessment and, upon request, to competent authorities.
In the two above cases, the PSUR should at least include reactive data regarding product complaints, reporting of serious incidents, FSCA and trend reports as documented in the system used to record the data and relevant data from literature research and relevant databases.
A workflow to determine whether a PSUR is required or not is provided with figure 1:
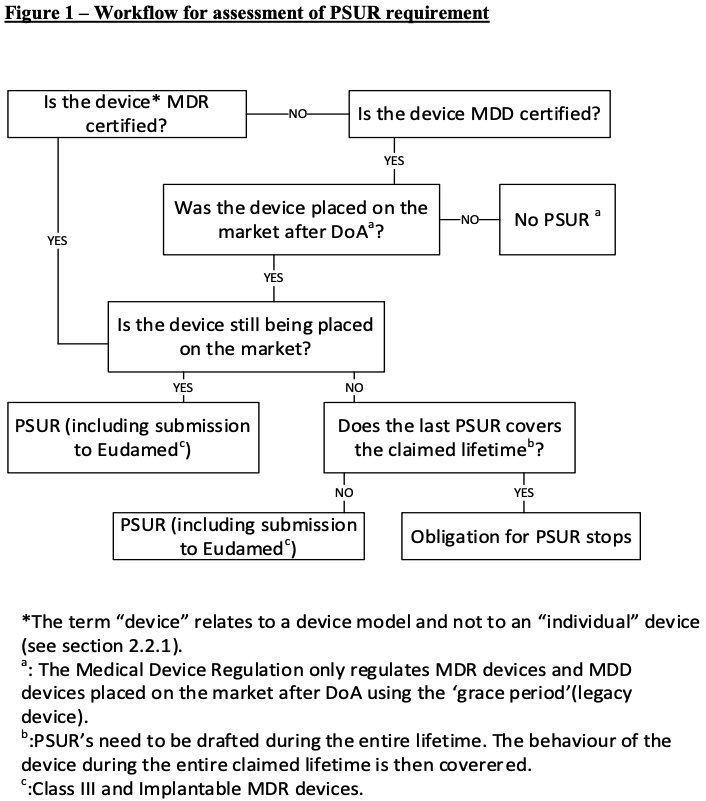
4 Grouping of devices
A PSUR should be drawn up per device and, where relevant, for a category or group of devices. If it covers a single device, it should be understood as the device associated with one Basic UDI-DI (or one device for legacy devices). Every device may include different variants or sizes.
According to Article 86(1), a same PSUR may cover a “category or group of devices”. As a consequence, multiple Basic UDI-DIs (or device families) may be covered in one PSUR.
Nevertheless the manufacturer should demonstrate the relevance of the grouping of several devices within the same PSUR by providing a justification.
4.1 General aspects for all PSURs
- In case a PSUR includes several Basic UDI-DIs, the data should be presented in a clear, organized manner so that it is easy to determine how each device performs independently.
- In case of a group of devices covered by the same PSUR, the manufacturer should assign a “leading device” which drives the schedule of that PSUR. The “leading device” needs to be the highest risk class or one of the highest risk class.
- The “leading device” determines the schedule applicable to the whole group of devices (data collection period covered, PSUR frequency, issuance timeline, PSUR reporting through EUDAMED or not).
- Therefore, for the other devices, these requirements should be aligned on the “leading device”, irrespective of their device class or certification date.
- When a device grouping has been established, it could be amended for the PSUR update(s) by removing or adding devices except for the “leading device” which cannot be changed.
- In case of a change related to the “leading device” (new device model /change of the Basic UDI DI), a new PSUR should then be issued.
- PSUR updates for the group of devices which includes the former “leading device” should continue in parallel independently it continues or not to be placed on the market.
- Due to the involvement of only one Notified Body for the review or evaluation of a PSUR, the grouping of devices within one PSUR is only possible for devices for which the conformity assessment activities have been carried out by the same Notified Body.
- In case a device or, where applicable, the leading device, is on the market with subsequent certificates of different notified bodies, the revision history provided should make a reference to the previous PSUR versions where the former Notified Body(ies) were involved and, when applicable, indicates the actions required or taken by previous Notified Body(ies).
4.2 Specific aspects for PSURs to be submitted in EUDAMED
- At least one Basic UDI-DI of class III or implantable devices needs to be referenced within a PSUR. A manufacturer can add several Basic UDI-DIs in case of grouping of devices.
- For the purpose of EUDAMED, every class III or implantable device should have at least one linked PSUR.
- For a same reporting period, a Basic UDI-DI can be referenced only in one PSUR as “leading device” but can be referenced in other PSURs provided this device is non-leading in those PSURs.
- When registering a PSUR in EUDAMED, the manufacturer should capture the Basic UDI-DIs of the class III or implantable devices belonging to the group via the EUDAMED web interface (see PSUR Web form in Annex V of this guidance).
- If the PSUR contains devices which are not Class III or implantable devices, as these devices do not require a PSUR in EUDAMED, the manufacturer should not capture them via the Web interface but list them in the body of the PSUR report as part of the PSUR scope.
- If the PSUR contains class III or implantable legacy devices, as these devices do not require (8) a PSUR in EUDAMED, the manufacturer should not capture them via the web interface but list them in the body of the PSUR report as part of the PSUR scope.
5 PSUR preparation and issuance
The manufacturer is responsible for preparing and updating the PSURs and making them part of the technical documentation as specified in Annex III of the MDR and in Annex XIII for custom-made devices (CMD).
5.1 Data collection period, issuance timeline, submission and schedule of PSURs
The data collection period should start at the device MDR certification date. If the device is not MDR-certified, the data collection period starts at MDR Date of Application (26 May 2021).
Note 1: for the first PSUR, the data analysis may be supported by the device’s historical data collected through the Post Market Surveillance activities as they were conducted prior to Date of Application or MDR Device Certification date (see section 5.2.2.1).
Note 2: The first PSUR may not cover an exact data collection period of 12 or 24 months. The first PSUR update(s) may then cover a period different than 12 or 24 months of post-market data to allow for data collection periods being contiguous to avoid any gap or overlap of data, i.e. the end of data collection period for one PSUR marks the start date of the next PSUR’s data collection period.
- PSUR preparation and issuance timeline
The PSUR preparation and issuance timeline refers to the period required for the manufacturer to prepare and submit or make available the PSUR after the end of the data collection period.
- PSUR submission / issuance
Depending on the class of the device and in accordance with Article 86(2), the manufacturer should either submit the PSUR to the Notified Body via EUDAMED or make it available to the Notified Body involved in the conformity assessment.
For those PSUR requiring submission through EUDAMED, the submission should occur with due diligence after the end of the issuance timeline (see section 5.2.1).
Schedule for PSUR updates
- The schedule is the generated cycle for (i) the start and the end of the data collection period covered by each PSUR and (ii) preparing and submitting the PSUR or making it available after the end of the data collection period.
Continuity of the schedule when a legacy device becomes MDR certified
- When an MDD compliant device becomes certified under the MDR without significant change in the sense of Art 120(3), the initial PSUR schedule established under the Article 120(3) regime may continue (unless otherwise agreed between the manufacturer and its Notified Body). Therefore, the schedule for the MDR device may not align on the initial MDR certification date.
- When a significant change in the sense of Article 120(3) occurs, the device should be considered as a new device. Based on the MDR certification date of this device, a new schedule needs to be started (unless otherwise agreed between the manufacturer and its Notified Body).
5.2 Main scenarios
Several scenarios should be considered depending on (i) the device has been directly MDR certified or (ii) is a legacy device which has become MDR certified during the transition period or (iii) is a legacy device which would remain an AIMDD or MDD device until the end of the transition period.
5.2.1 New Devices certified under MDR (not previously marketed or put into service under AIMDD 90/385/EEC & MDD 90/42/EEC)
The requirement to issue a PSUR applies from the date the device has been certified under MDR (see definition of “device certification date” in section 1.2).
Data collection period
The starting point for the data collection period is based on the first device certification date for the MDR device.
The end of the data collection period of the PSUR should be aligned to the anniversary date of the issued MDR certificate or in agreed schedule with the Notified Body.
The defined schedule for PSUR updates should continue as planned for any future recertification and remain aligned to first certificate issued under the MDR.
In exceptional circumstances, this date may be changed on alignment between the Notified Body and the manufacturer and this change should be justified by the manufacturer in the first available PSUR. The data collection periods should be contiguous: this may lead to a shorter or longer data collection period covered by the next PSUR which results from these exceptional circumstances.
PSUR preparation and issuance / submission
- For class III devices or implantable devices
Manufacturers should submit PSURs through EUDAMED to the Notified Body involved in the conformity assessment.
The PSUR should be submitted in EUDAMED from the end of the data collection period.
- For non-implantable devices of either class IIa or class IIb
The PSUR is not submitted to EUDAMED: manufacturers should make PSURs available to the Notified Body involved in the conformity assessment and, upon request, to the Competent Authorities.
5.2.2 Legacy devices which become certified under MDR during the transition period (previously marketed or put into service under AIMDD 90/385/EEC or MDD 93/42/EEC)
It should be noted that “legacy devices” are subject to the requirements laid down in Article 86 based on their classification in accordance with the MDD. A possible change of their risk class under the MDR should not be taken into account during the transition period. Active implantable devices subject to the AIMDD should be considered as class III devices for the purpose of applying the PSUR requirement during the transition period (9).
For “legacy devices”, manufacturers should not submit the PSUR in EUDAMED but make it available upon request to the Competent Authorities and the Notified Body.
5.2.2.1 An initial PSUR has not been issued for the MDD compliant device
Specific considerations are listed below:
- When a “legacy device” becomes certified under the MDR and no PSUR has been issued by the time of certification (e.g. the MDR certification occurs within the first (second) year after MDR DoA), the PSUR should be prepared at the anniversary date of the MDR certification (or issuance date of the Declaration of Conformity (DoC)) or in alignment with the Notified Body.
- When relevant and for the first PSUR for the MDR device, it is recommended that the data analysis be supported by the device’s “historical data” collected through the Post Market Surveillance activities conducted prior to the Date of Application or MDR Device Certification date. The “historical data” may be presented in a different format than the MDR ones and could be limited to a summary.
5.2.2.2 An initial PSUR was issued for the legacy device before it becomes MDR certified
Data collection period
– First PSUR
The data collection period for the MDR certified device should start at the end of data collection period for the PSUR drawn up for a “legacy device” in accordance with Article 120(3) MDR.
There is no need to consider the data before MDR date of application since a PSUR for the corresponding legacy device has already been issued.
5.2.3 Legacy devices which do not become MDR certified during the transition period
Manufacturers should follow the rules corresponding to the class of the valid MDD certificate. It should be noted that the Notified Body (10) which provided the certification of legacy devices under MDD or AIMDD remains responsible for the appropriate surveillance until the end of the transition period or the withdrawal of the MDD or AIMDD certificate if it occurs before the end of the transition period.
Data collection period
The data collection period for the first PSUR should start at the MDR DoA.
However, when relevant and for the first PSUR, it is recommended that the analysis be supported by the device’s historical data collected through the Post Market Surveillance activities as they were conducted prior to the MDR DoA. The data related to time periods prior to MDR DoA may be presented in a different format and could be limited to a summary.
PSUR issuance / submission
Manufacturers should not submit the PSUR in EUDAMED but make PSURs available upon request to Competent Authorities and the Notified Body.
5.3 Specific provisions for custom-made devices
- The preparation of the PSUR is only required for classes IIa, IIb and class III custom-made devices complying with the requirements of the MDR, per schedule in accordance with device class as described in Article 86 and in accordance with guidance MDCG 2021-3 for custom-made devices (11).
- All custom-made devices which have been put on the market from MDR DoA are MDR devices: therefore no legacy devices need to be considered.
- For new MDR devices, the first PSUR should be prepared within one (class IIb and III) or two (class IIa devices) year(s) following the first Statement of Conformity for this type of devices is issued. For these devices, the PSUR is not submitted to EUDAMED but it should be made available to the Competent Authorities upon request.
- The PSUR may cover entire groups or families of custom-made devices (e.g., hips, knees, shoulders, extremities). A justification should always be provided by the manufacturer for the grouping of several devices and device families within the same PSUR.
- In case of Class III implantable custom-made devices, PSURs are not required to be sent to notified bodies but should be part of the documentation (see MDCG 2021-3) according to Section 2 of Annex XIII of the MDR.
A summary table of the applicable requirements for the various device classes for MDR devices and legacy devices is provided in Annex IV of this guidance.
Decision trees based on section 5 are provided within figures 2 and 3.
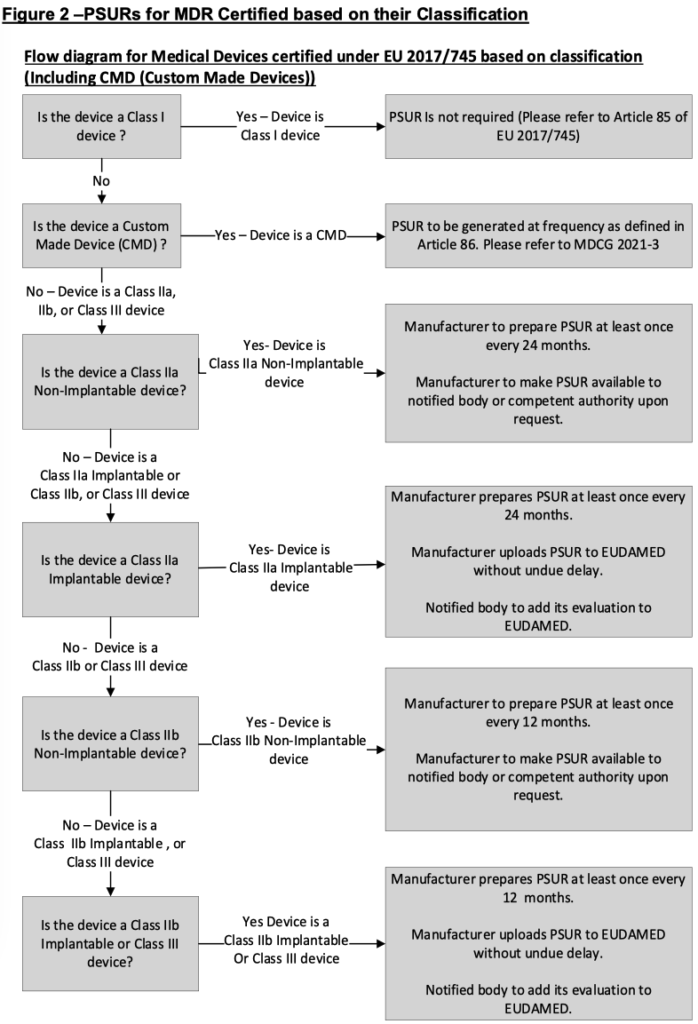
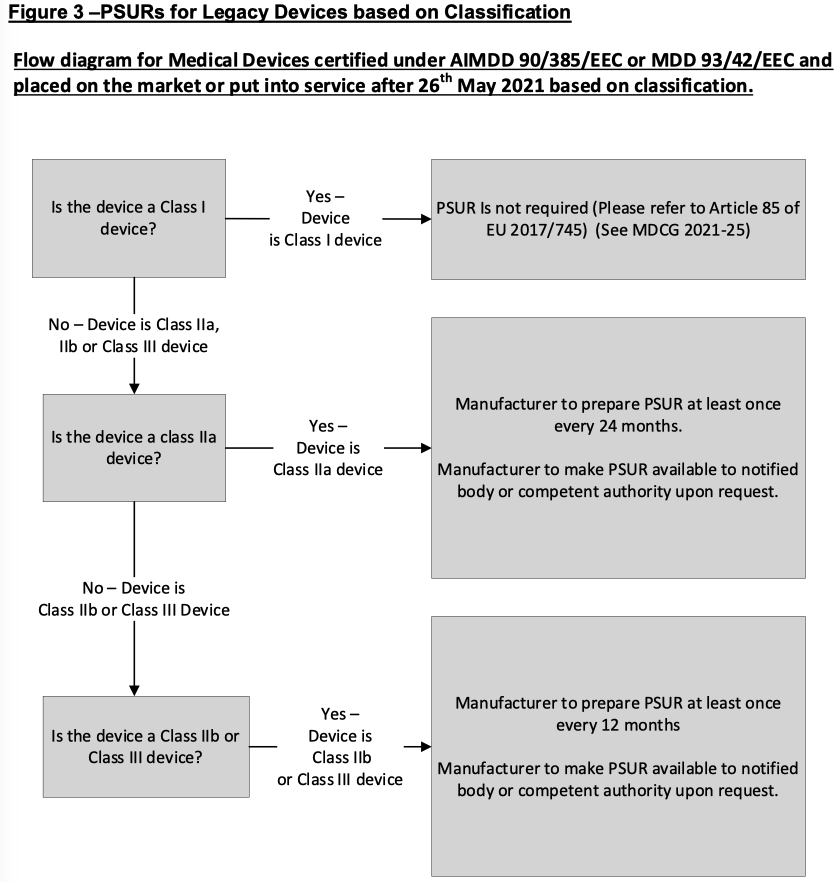
5.4 PSUR submission by the manufacturer in the absence of EUDAMED
Until EUDAMED becomes fully functional, manufacturers or their authorised representatives should apply the respective national provisions and take into account MDCG 2021-1 Rev.1 (12)
For class III or implantable devices of either class IIa or class IIb which are MDR certified, manufacturers should, in the absence of EUDAMED, deliver the PSURs to the relevant notified bodies by appropriate means. The manufacturer should align with their Notified Body on the submission method. For Competent Authorities, the manufacturer should provide the PSUR upon request.
ANNEX I: Template for the PSUR
The PSUR should be generated as a stand-alone document that can be assessed independently from the supporting documentation. The PSUR should provide a general overview of all post-market surveillance activities and the data collected and analysed based on the PMS plan for the device. Therefore, the aim of the PSUR is not to duplicate all data and reports generated by the PMS Plan but it should summarize all results and conclusions.
The manufacturer should specify the relevant information and sections of the different reports and provide a summary of the data collected, their assessment and conclusion as well as any actions taken when appropriate. If a manufacturer decides that specific data sets are not used or deemed to be not required, the manufacturer should duly justify the absence of the data sets not included in the relevant sections of the PSUR.
It is recommended to add an executive summary in particular as regards the main relevant information related to benefits and risks and to the changes in the acceptability of the benefit-risk profile.
PSUR cover page
The PSUR cover page includes the relevant data to allow distinguishing between the various PSUR updates.
For those PSURs submitted to EUDAMED, the PSUR form (EUDAMED Web form, see Annex V) can be considered as a cover page. A Table of Contents should also be present for all PSURs.
The cover page should at least include the following information:
- Manufacturer information
- Medical device(s) covered by the PSUR
- Notified body name and organization number;
- PSUR reference number assigned by the manufacturer*;
- Version number of the PSUR;
- The data collection period covered by the PSUR;
- Table of contents.
Executive summary
When an executive summary is produced, it should provide a brief overview of the PSUR content and an overall conclusion in relation to the benefit-risk determination.
It should include the following information:
- A brief description and status of actions taken by the manufacturer based on the previous PSUR;
- A brief description and status of actions taken by the Notified Body as part of the review of the previous PSUR;
- In case the data collection period is changed by the manufacturer, a justification should be provided, and a statement should be provided whether the change affects the comparability of the results gained;
- Once the conclusions of the PSUR have been completed, the main results of the current PSUR should include a clear and bold statement declaring whether the benefit-risk profile has been impacted, negatively or positively or remains unchanged, based on the information reported within the current PSUR. The statement could be a simple expression, for example “Based on the analysis of the collected data, it is concluded that the benefit-risk profile of the device(s) has not been (or has been) adversely impacted / remains unchanged”.
Description of the devices covered by the PSUR and their intended uses (Article 86.1)
This section is intended to provide an overview of the devices covered by the PSUR and the possible changes to its scope. The added and removed devices should be clearly identified. The following information should be included for the devices covered by the PSUR:
- Device Classification (risk class of device) in accordance with the applicable classification rules.
- Date from one of the following: first declaration of conformity, first EC / EU Certificate issued, first date device CE-marked, first placed on the market, first put into service, if software, date first made available.
- Status of the device(s): on the market, no longer placed on the market, recalled, field safety corrective action initiated.
- The intended purpose of the device(s) as per the Instructions for Use according to Annex I, Chapter III, 23.4(b) MDR, any indications, contra-indications, and target populations.
– For MDR Devices
- The information shall be broken down by the Basic UDI-DI(s) and explain any device changes within each Basic UDI-DI compared to the previous PSUR to comprehend possible changes in results compared to the previous PSURs.
- Provide the device trade name(s) associated to the corresponding Basic UDI- DI(s) and the European Medical Device Nomenclature (EMDN).
– For Legacy Devices
- The information shall be broken down by device group/family of devices
- Provide device trade name(s) (this includes all trade names the device may have on the market in different Member States) and European Medical device Nomenclature (EMDN).
– For Custom-Made Devices (MDR)
- Provide the required information by device group.
Grouping of the Devices
Information regarding the grouping of devices are provided in section 4 of this guidance.
- The manufacturer should justify the grouping of the devices in one PSUR.
- The justification could be based on the benefits to report multiple devices in one PSUR or alternatively the disadvantages to report each device in separate PSURs.
- In case the group of devices is changed, a justification for the change should be provided. The manufacturer should also provide the PSUR reference number of the PSUR where the data of the removed device(s) are reported.
- The manufacturer should define the “leading device” according to which the PSUR schedule is determined.
- The PSUR reference number is attached to the “leading device” and should remain unchanged for the PSUR updates, provided the “leading device” within the grouped devices has remained the same.
Volume of Sales (Article 86.1)
- The manufacturer should consider all the devices placed on the market. This could be volumes of sales, units shipped, or units implanted or another suitable indicator. Whichever method is used should be consistent throughout the PSUR in all areas to allow for a comparison of data. Provide accurate information the number of devices sold. The data should be presented by year to year (see Annex II, Table 1).
- Provide further information on the volume of sales in respect to the various sizes, models and configurations of the device as deemed necessary.
- Indicate to what criteria the number of devices on the market is provided:
- Devices placed on the market or put into service;
- Units distributed within each time period;
- Number of episodes of use (for reusable devices);
- Active installed base;
- Units distributed from the date of declaration of conformity or EC/EU mark approval to the end date of each time period;
- Number of devices implanted;
- Other – description/rational should be provided.
Size and other characteristics of the population using the device (Article 86.1)
- Evaluate how many patients have been exposed to the device and the characteristics of the exposed patient group(s).
- Estimate the number of patients exposed, as the sales numbers alone do not necessarily reflect the number of uses of the device (usage frequency). There are different scenarios as: Active devices may have a lifetime of several years with multiple uses each day, resulting in high number of patients exposed to the device (e.g. CTs). In case of implants, multiple devices may be used in one patient, e.g. several bone screws in one surgery. For other devices, the sales numbers directly correlate with the patient number exposed to the device.
- Describe the usage of the device in different patient populations and when available compare it to the expected usage and identify the possible over-represented or under-represented patient groups if clinically relevant and known by the manufacturer.
- When possible, consideration should be given to patient demographic aspects.
- When applicable, evaluate the effect of the detected changes to findings obtained previously and in the current PSUR.
Post-Market Surveillance : Vigilance and CAPA information
Background information should be gathered prior to the current PSUR and may include, for example, the achieved safety and performance of the device, information related to intended benefits achieved or not and description of new risks or emerging trends reported in earlier PSURs.
Vigilance data consist of information concerning serious incidents, field safety corrective actions (FSCAs) and trend reports. The data could be presented in tables, figures and/or in text format. The aim of the data presentation is to provide an accurate summary and appraisal of the Vigilance data (Article 87 and Article 88 MDR) and CAPA data (Article 83(4) and Article 86 MDR) for the reported data collection period and to compare with the same types of data from the previous PSURs.
The data should be presented by the device (Basic UDI-DI), device group (CMD) or device group/family level (legacy devices). When justified, the data can be presented for combinations of devices, for example, a device and its accessory.
a) Information concerning Serious Incidents (Article 87, Annex III MDR)
- The aim is to present the serious incidents and their impact on the overall device safety. This section should characterize the data from at least three different perspectives: the device problems, the root cause and the health effects on the person(s) affected. In addition to the data, provide a summary text regarding any new types of serious incidents which have occurred since the last report.
- Data regarding serious incidents should be reported using the IMDRF Adverse Event Terminology (AET) (13), when available. With regard to the historical data, the usage of the IMDRF Adverse Event Terminology is not required.
- The usages of the Level 2 terms/codes are considered sufficient to enable the grouping of the serious incidents;
- Report both the codes and the terms.
- When applicable report both absolute figures and rate of the serious incidents and split the data by region (EEA+TR+XI) and worldwide.
- Examples of the data presentation are shared in Annex II of this guidance.
- The most frequent medical device problems by IMDRF Adverse Event Terminology Annex A – “Medical Device Problem”, by year to year- (see Annex II, Table 4).
- The most common investigation findings as part of the completed “cause investigation” of the serious incidents by IMDRF Adverse Event Terminology Annex C – “Investigation Findings”, (see Annex II, Table 5).
- The health impacts on the person affected as a consequence of the medical device serious incident by IMDRF Adverse Event Terminology Annex F – “Health Impact”, including the term and code. It could also be used for the 4-year summary data (starting as of the device MDR certification date or the MDR date of application for legacy devices) and split the data by the IMDRF Adverse Event Terminology Annex D – “Investigation Conclusion” (including term and code). Use only the most relevant investigation conclusion terms/codes which are related to the detected health impacts. Report the most common health impacts as well as any cases resulting into death, regardless if they are included in the most common health impacts. In addition, split the data by region (see Annex II, Table 6).
b) Information from Trend Reporting (Article 88, Annex III MDR, non-serious incidents and expected undesirable side effects)
The data related to the trend reports will be detailed after the adoption of the MDCG guidance on trend reporting.
c) Information from Field Safety Corrective Actions (FSCA) (Article 87, Annex II MDR)
- Provide a summary of the FSCAs for the period of the PSUR and compare with the information from the previous PSURs.
- The summary should include the following information (14):
- types of actions.
- issuing date,
- scope of the FSCA,
- status of the FSCA at the time of the PSUR,
- manufacturer’s reference number,
- a brief description of the reason for action and description of action and impacted regions.
An example of the data presentation is presented in Annex II of this guidance (table 7).
d) Preventive and / or Corrective Actions (CAPA) (Article 83.4 and Article 86 MDR)
- Provide a list of all preventive and / or corrective actions (CAPA) according to Article 83(4) and to Article 86.
- The following information should be provided for each CAPA:
- the type of action,
- initiation date,
- scope of the CAPA,
- status of the action,
- manufacturer’s reference number,
- CAPA description,
- the root cause (internal codes with the explanation, IMDRF terms/codes or free text),
- effectiveness of the CAPA
An example of the data presentation is presented in Annex II of this guidance (table 8).
Post-Market Surveillance: information including general Post-Market Clinical Follow-up (PMCF) information (Annex III and Annex XIV, Part B, 6.2(a) and (f) MDR)
The data that should be reported in this section consist of other PMS datasets not referred to above and are generated by general methods and procedures of PMCF (Annexes III, Annex XIV Part B, 6.2 (a) and (f) MDR). The sections below should be completed in alignment with the PMS and PMCF plans.
A list of collected data from other sources of clinical data in the post-market phase should be provided. Safety and performance data generated from these activities should be used also for comparison to other similar devices with the same intended purpose.
a) Feedbacks and complaints from users, distributors and importers
- All feedback from users, distributors and importers and complaints not reported in the Vigilance section above should be considered in this section. The most common complaints should be presented within this section of the PSUR with the following considerations:
- Grouping of complaints by IMDRF Adverse Event Terminology Annex A –“Medical Device Problem” (including the term and code) or internal event codes including term;
- Occurence rate (including reference chosen);
- Justification for inclusion of these groups of complaints and exclusion of those not presented;
- Information whether the presented complaints have led to initiation of preventive and / or corrective actions (CAPA).
b) Scientific Literature Review of relevant specialist or technical literature
- For detailed information about literature searches conducted and results generated, the manufacturer may refer to the technical documentation.
c) Public Databases and /or Registry Data
- Provide a list of all registries reviewed including the following information: the name or registry reference, type of registry (Prospective or Retrospective data collection);
- Provide a list of findings in comparison to the devices with same intended use and justify any identified differences. Provide information about any new risks identified from this data set.
d) Publicly Available Information about Similar Medical Devices
- Additional publicly available information may include information gathered from other manufacturers of similar medical devices, (e.g. results of a manufacturer’s specific PMCF study made publicly available in the manufacturer’s Summary of Safety and Clinical Performance (SSCP), Cochrane Library or other libraries);
- The type and location of this information should be provided, and when possible a comparison of the devices with same intended purpose should be evaluated with any possible differences in safety and performance reported.
e) Other Data Sources
- The other used data sources could be for example real-world data from electronic health records and digital health-monitoring devices;
- Provide a list of the used data sources and findings with specific reference to safety and performance of the device.
Specific Post-Market Clinical Follow-up (PMCF) Information (Article 86, MDR Annex XIV, Part B, 6.2(b))
This section should include a summary of the findings generated from the analysis of specific PMCF activities performed by the manufacturer as defined in Annex XIV, Part B, 6.2(b). This section is not limited to PMCF studies and should include other specific PMCF activities conducted by the manufacturer.
For this section, the manufacturer should refer to the main findings of the PMCF and, when available, to the conclusions documented in the PMCF Evaluation Report to allow for a comprehensive assessment of the specific PMCF activities it has performed.
Summary of Findings and Conclusions of the PSUR
In this section of the PSUR, the manufacturer should consider the validity of the collected data taking into consideration any deficiencies or bias, and provide a conclusion on the benefits and risks of the device from the gathered data. In the case when these data have had any impact on the overall benefit-risk determination, this should be described. The manufacturer should also outline all actions that have been taken as a result of the analysis of data collected since the last PSUR.
a) Validity of the collected data
- The manufacturer should identify any limitations to the data that have been collected, this could include for example reduced sales or usage of the device, known bias from feedback obtained or enrolment into a PMCF study.
- The manufacturer should consider whether these limitations impact the ability to formulate meaningful conclusions and whether an impact assessment of the overall benefit-risk profile is still possible.
b) Overall conclusions from the analysis of the collected data
- The manufacturer should outline any new or emerging risks identified or when common occurrences of poor performance or claimed benefits have not been achieved within the current reporting period. When there are new or emerging risks that have been identified, the manufacturer should consider any specific patient groups, device models, accessories used, geographical regions impacted, duration of risk etc. Specific information should be provided on the seriousness and the full potential clinical impact of these risks.
- The manufacturer may also describe any new benefits that have been identified from the reporting period.
- The manufacturer should formulate evidence-based conclusions to determine whether the benefit-risk profile of the device has changed or not.
- Finally, within the conclusion, the manufacturer should declare whether there has been an adverse impact on the benefit-risk profile of the device or the benefit-risk profile remains unchanged.
c) Actions taken by the manufacturer
- The manufacturer should describe any specific actions that have been taken to address any newly identified or emerging risks and occurrences of poor performance.
- The manufacturer should identify all actions initiated during the data collection period as described in Article 83 (3).
ANNEX II: Templates for the Presentation of Data in the PSUR
These tables are intended to provide guidance to manufacturer and are only examples. It is up to the manufacturer to present the data in the most appropriate manner depending on the nature of the data and of the device. Please read this Annex II in conjunction with Annex I when forming tables.
Table 1. Volume of sales* by region over time
| Basic UDI-DI/ Legacy device name or model | |||||
|---|---|---|---|---|---|
| Total Number of devices | Reporting Day+ preceding 12 months (N) | N – 12 months (N2) | N2-12 months (N3) | N3-12 months (N4) | |
| EEA+TR + XI** | |||||
| Worldwide | |||||
* Indicate according to which criteria the number of devices on the market is provided (Annex II, 4.1)
** EEA: European Economic Area, TR: Turkey, XI: Northern Ireland.
Table 2. Estimated size of the population using the device* over time
| Estimated size of population using the device Reporting Day+ preceding 12 months (N) | Estimated size of population using the device N – 12 months (N2) | Estimated size of population using the device N2-12 months (N3) | Estimated size of population using the device N3-12 months (N4) | |
|---|---|---|---|---|
| EEA+TR + XI | ||||
| Worldwide |
* When clinically relevant and known by the manufacturer
Table 3. Characteristics of the population using the device* over time
| Characteristic X of population using the device Reporting Day+ preceding 12 months (N) | Characteristic X of population using the device N – 12 months (N2) | Characteristic X of population using the device N2-12 months (N3) | Characteristic X of population using the device N3-12 months (N4) | |
|---|---|---|---|---|
| EEA+TR + XI | ||||
| Worldwide |
* Characteristics of the population using the device is defined by the manufacture based on the usage of device
Table 4. Total number (N) and rate (%)*of the serious incidents by IMDRF Adverse Event Terminology (AET) Annex A – Medical Device Problem by time and region over time
| Basic UDI-DI/Legacy Device name or model | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| IMDRF Adverse Event – Medical Device Problem code (Annex A) and term by region | Reporting Day+ preceding 12 months (N) | N – 12 months (N2) | N2-12 months (N3) | N3-12 months (N4) | |||||
| N | % | N | % | N | % | N | % | ||
| EEA+TR + XI | |||||||||
| Worldwide | |||||||||
| EEA+TR + XI | |||||||||
| Worldwide | |||||||||
*The denominator is compatible to the number of devices in table 1 or based on manufacturer’s reasoning e.g. reusable instruments
Table 5. Total number (N) and rate (%)* and of the serious incidents by IMDRF AET Annex C – Cause Investigation-Investigation Findings by time and region over time
| Basic UDI-DI/Legacy Device name or model | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| IMDRF Adverse Event – Investigation Findings (Annex C) code and term by region | Reporting Day+ preceding 12 months (N) | N – 12 months (N2) | N2-12 months (N3) | N3-12 months (N4) | |||||
| N | % | N | % | N | % | N | % | ||
| EEA+TR +XI | |||||||||
| Worldwide | |||||||||
| EEA+TR +XI | |||||||||
| Worldwide | |||||||||
* The denominator is compatible to the number of devices in table 1
Table 6. IMDRF AET Annex F – Health Effects-Health Impact code of the serious incidents by IMDRF Adverse Event Terminology Annex D – Investigation Conclusion in last 4-years
BASIC UDI-DI/Legacy Device name or model | ||||||
|---|---|---|---|---|---|---|
IMDRF Adverse Event Health Impact (Annex F) code and term by region | Number of serious incidents | Investigation conclusion code+ term 1 % | Investigation conclusion code+ term2 % | Investigation conclusion code + term3 % | Investigation conclusion code + term4 % | |
EEA+TR + XI | ||||||
Worldwide | ||||||
EEA+TR + XI | ||||||
Worldwide | ||||||
Table 7. FSCA initiated in current reporting period and open FSCAs *
Type of action | Issuing date | Scope of the FSCA | Status of the FSCA** | Manufacturer Reference number | Rationale and description of action taken | Impacted regions |
|---|---|---|---|---|---|---|
* Will be further developed when the new FSCA form is in use
**follow-up, final at the time the data collection time ended
Table 8. CAPA initiated in current reporting period and open CAPA
BASIC UDI-DI/Legacy Device name or model | |||||||
|---|---|---|---|---|---|---|---|
Type of action | Initiation Date | Scope of the CAPA | Status of the CAPA | Manufacturer Reference number | CAPA description | Root cause* | Effectiveness of the CAPA if closed** |
*internal codes with the explanation, IMDRF codes or free text
**If CAPA is still open then this is not applicable, if CAPA is closed comment on whether it is resolved, not resolved or comment if additional CAPA has been opened.
ANNEX III: General Information Related to the Presentation and Assessment of the Collected Data by the Manufacturer
1. How Data Should be presented
- Each dataset specified in the PMS Plan should be presented and analysed individually. A summary providing the used datasets including the PMCF data should highlight the limitations related to the collected data.
- Datasets should be split by Basic UDI-DI or model of the device if the Basic UDI-DI does not exist.
- If the group of devices or the devices within a Basic UDI-DI(s) have changed then it is necessary to report separately the data with former and later combination of devices.
- The data should be split by region, when applicable. The used regions are EEA, TR, XI and worldwide. Worldwide data include data from EEA, TR and XI.
- Each PSUR should contain data from the data collection period of this PSUR compared with the same types of data from the previous PSUR periods (see tables 2 to 5 of Annex II of this guidance).
- The first PSUR data collection is not retrospective (does not go before the date of application of the MDR) except when no PSUR has been issued for the corresponding MDD compliant device: it is then recommended that the “historical data” collected before the MDR DoA be considered for the first PSUR of the MDR compliant device (see section 5.2.2.1).
- Data should be reported by year to year:
- Class III and Class IIb: Reporting Day+ preceding 12 months (N); N – 12 months (N2); N2-12 months (N3); N3-12 months (N4)
- Class IIa: Reporting Day+ preceding 24 months (N); N – 24 months (N2)
- The manufacturer should present the data utilizing the International Medical Device Regulators Forum (IMDRF) Adverse Event Terminology when the content of the data facilitates it.
- The following IMDRF Adverse Event Terminologies, terms and codes should at least be utilized:
- Annex A: Medical Device Problem
- Annex C: Cause Investigation – Investigation Findings
- Annex D: Cause Investigation – Investigation Conclusion
- Annex F: Health Effects – Health Impact
- Level 2 terms are satisfactory to enable the grouping of cases.
- When the Level 2 terms are not available, manufacturers can use Level 1 terms/codes.
2. How Data Should be assessed by the manufacturer
- Findings from all datasets should be considered and evaluated by comparing data from the various sources and identifying possible conflicting results.
- The manufacturer should assess the results considering the different patient populations, sizes and models of the device, device combination or variants. When applicable, the manufacturer should evaluate the findings in relation to the state of the art.
- The manufacturer should assess the data in relation to the thresholds concerning known risks and side effects and benefits intended to be gained.
- The manufacturer should identify factors that support or refute previously identified safety and performance concerns as well as evidence relating to new safety signals or emerging risks.
- The manufacturer may also describe any new benefits that have been identified from the reporting period and benefits not achieved as intended.
- When applicable, the IMDRF Terminologies for Categorized Adverse Event Reporting should be used in the analysis.
- If the device is used in a combination of devices, the analysis should identify the role of each device in comparison to other devices or accessories used together.
- The performance and safety of the device should be compared to other devices with the same intended use.
ANNEX IV: PSUR Requirements – Summary Table for MDR and Legacy Devices
MDR devices
| MDR certified Class III or IIb Implantable devices | MDR certified Class IIa Implantable devices | MDR certified Class IIb Non – Implantable devices | MDR certified Class II a Non – Implantable devices | Custom-made devices | |
|---|---|---|---|---|---|
Frequency (at least) | Annually | Every 2 years | Annually | Every 2 years | Same as other devices according their MDR risk classification |
| PSUR uploaded to EUDAMED | Yes | Yes | No | No | No |
| PSUR (first) availability | One year after device certification date. For newly certified devices, a PSUR is not required at the time of initial certification | 2 years after device certification date. For newly certified devices, a PSUR is not required at the time of initial certification | One year after device certification date. For newly certified devices, a PSUR is not required at the time of initial certification | 2 years after device certification date. For newly certified devices, a PSUR is not required at the time of initial certification | One year (class IIb and III) or two years (class IIa devices) when the first Statement of Conformity (SoC) for custom-made devices is issued |
| PSUR preparation | The manufacturer should prepare and submit the PSUR by means of Eudamed after the end of data collection period. | The manufacturer should prepare and submit the PSUR by means of Eudamed after the end of data collection period. | Manufacturers to make the PSUR available during notified body’s surveillance activities | Manufacturers to make the PSUR available during notified body’s surveillance activities | Manufacturers to make the PSUR available upon request |
| Notified Body availability | PSUR made available by the manufacturer via EUDAMED | PSUR made available by the manufacturer via EUDAMED | PSUR made available by the manufacturer during surveillance activities | PSUR made available by the manufacturer during surveillance activities | N/A |
| Competent Authorities | Made available via EUDAMED | Made available via EUDAMED | Made available upon request | Made available upon request | Made available upon request |
| In Technical Documentation | Yes – part of the technical documentation as specified in Annexes II and III of MDR | Yes – part of the technical documentation as specified in Annexes II and III of MDR | Yes – part of the technical documentation as specified in Annexes II and III of MDR | Yes – part of the technical documentation as specified in Annexes II and III of MDR | In case of Class III implantable custom-made devices, PSURs must be part of the documentation for custom-made devices (see MDCG 2021-3) according to Section 2 of Annex XIII of the MDR |
| Data collection period | aligned on the device certification date or in agreed schedule with Notified Body | aligned to device certification date | |||
| End of PSUR requirement | the PSUR remains mandatory by the manufacturer until the end of the device lifetime defined in the technical documentation has been reached | ||||
Legacy devices
MDD certified Class III or IIb Implantable devices | MDD certified Class IIa Implantable devices | MDD certified Class IIb non – Implantable devices | MDD certified Class IIa non – Implantable devices | |
|---|---|---|---|---|
Frequency (at least) | Annually | Every 2 years | Annually | Every 2 years |
PSUR uploaded to EUDAMED | No | No | No | No |
PSUR (first) availability | During the course of the calendar year following MDR date of application | During the course of the second calendar year following MDR date of application | During the course of the calendar year following MDR date of application | During the course of the second calendar year following MDR date of application |
PSUR preparation | The PSUR should be prepared by the manufacturer and made available to the notified body during surveillance activities and, upon request, to competent authorities | |||
Notified Body availability | Manufacturers to make PSURs available during notified body’s surveillance activities | |||
Competent Authorities | Manufacturer to make PSUR available upon request | |||
In Technical Documentation | Yes – part of the technical documentation | |||
Data collection period | The data collection period should start at 26th May 2021 and contain one year of data | The data collection period should start at 26th May 2021 and contain two years of data | The data collection period should start at 26th May 2021 and contain one year of data | The data collection period should start at 26th May 2021 and contain two years of data |
End of PSUR requirement | If the last PSUR of MDD or AIMD device does not cover the lifetime of the device and the device is not certified under MDR (remains as a legacy device), then the issuance of the PSUR remains mandatory by the manufacturer, after the transition period until the end of the device lifetime defined in the technical documentation | |||
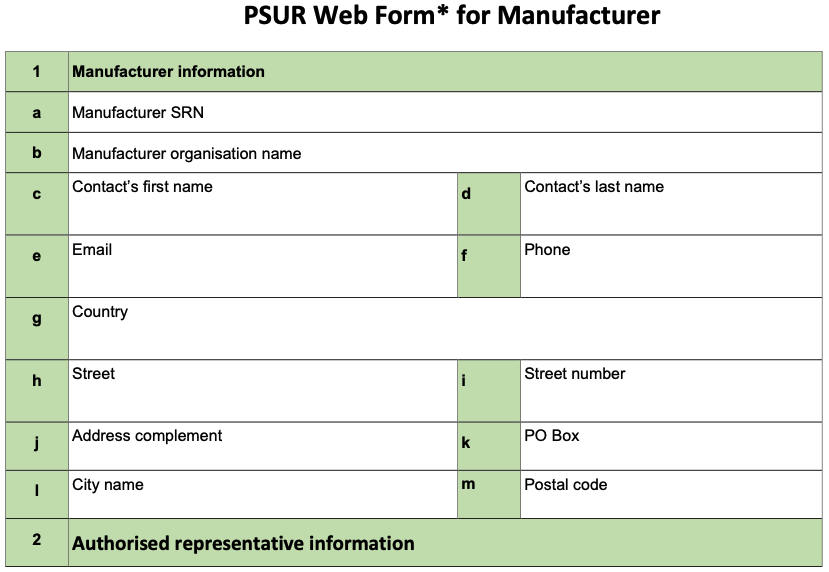
ANNEX V: PSUR Web Form for Manufacturer
- The PSUR Web form for manufacturer contains all the relevant administrative data necessary for the registration of the PSUR in EUDAMED: certain fields are automatically populated by EUDAMED e.g. Notified Body, Manufacturer, Single Registration Number (SRN) while other data need to be filled up by the manufacturer via EUDAMED Web interface.
- When EUDAMED becomes fully functional, the manufacturer should upload the PSUR in PDF format into EUDAMED for MDR class III devices or implantable devices and provide the information* of the PSUR Web form directly through the EUDAMED Web interface.
- The manufacturer should create a PSUR reference number which should remain the same for the PSUR updates. In case of grouping of devices within one PSUR, the PSUR reference number relates to the leading device.
- When registering a PSUR in EUDAMED, the manufacturer should capture the Basic UDI-DIs of all the Class III or implantable devices belonging to the group via the web interface.
- For PSURs which are not required in EUDAMED, the PSUR Web form is not applicable. Instead, the manufacturer should fill in the information required in the PSUR cover page (see Annex I of this guidance).
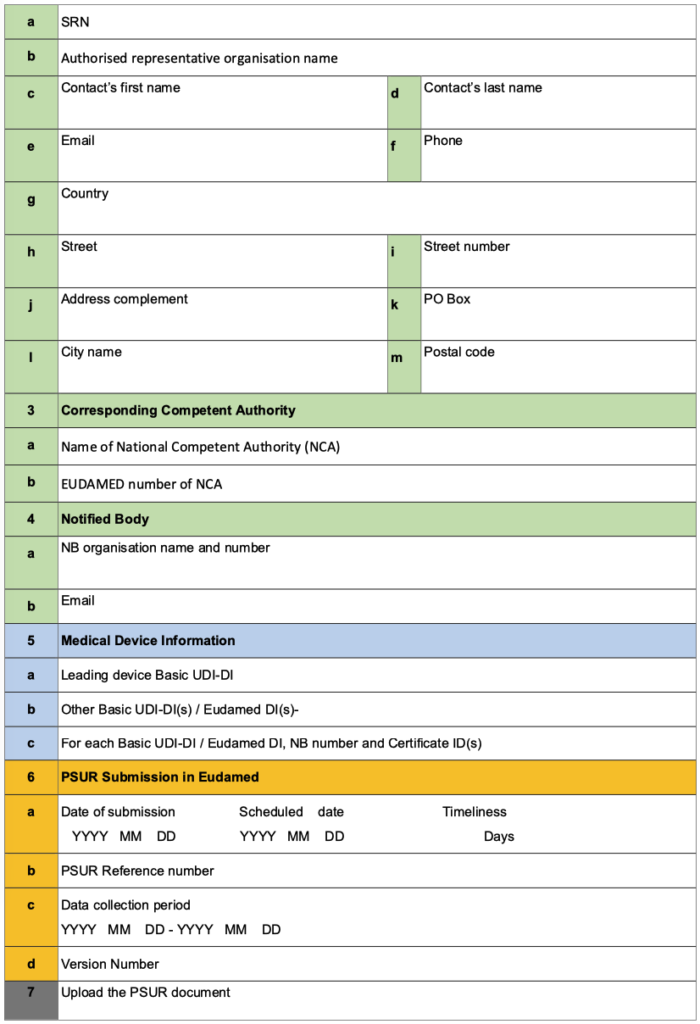
* Only the fields and content of the PSUR Web form need to be considered and not its structure which may be different in the EUDAMED web interface.
Footnotes
(1): MDR, Article 2(60)
(2): See point 11 of MDCG 2022-14 Position paper “Transition to the MDR and IVDR – Notified body capacity and availability of medical devices and IVDs”
(3): MDR, Article 2(3)
(4): MDCG guidance 2021-25, Regulation (EU) 2017/745 – application of MDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2021 in accordance with Directives 90/385/EEC or 93/42/EEC October 2021
(5): See section 2.2.1. first paragraph on the meaning of the term “device”.
(6): “If, in the course of the post-market surveillance, a need for preventive or corrective action or both is identified, the manufacturer shall implement the appropriate measures and inform the competent authorities concerned and, where applicable, the notified body”, Article 83(4) first sentence.
(7): MDR, Article 2(24)
(8): See MDCG guidance 2021-25 – Application of MDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2021
(9): See section III.1 last bullet point of MDCG guidance 2021-25 on application of MDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2021
(10): See MDCG guidance 2021-25 on legacy devices
(11): MDCG 2021-3 “Questions and Answers on Custom-Made Devices”
(12): MDCG guidance 2021-1 Rev.1:“Guidance on harmonised administrative practices and alternative technical solutions until EUDAMED is fully functional”
(13): Link to IMDRF website
(14): When EUDAMED will become fully functional, the information that need to be collected may change and the information presented in this section should be updated accordingly.