
MDCG 2022-2 Rev.0
Guidance on general principles of clinical evidence for In Vitro Diagnostic medical devices (IVDs)
Disclaimer: This document is an interactive version of the original MDCG document. We will keep it up-to-date.
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.
Table of Contents
1. Purpose
This document outlines the general principles of clinical evidence and provides guidance on the continuous process of performance evaluation for in vitro diagnostic medical devices (hereafter referred to as IVDs), as set out in Regulation (EU) 2017/746 – In Vitro Diagnostic Medical Device Regulation (IVDR).
This guidance describes the approach by which collection, generation and documentation of supporting data for an IVD may be conducted prior to the placing on the market or putting into service. As the performance evaluation will be updated throughout the life cycle of an IVD, this document also addresses principles related to post-market surveillance, such as post-market performance follow-up.
The target audience of this document is IVD manufacturers, investigators and study sponsors. This document is also intended to inform regulators, notified bodies and other stakeholders when considering clinical evidence provided by manufacturers.
This document has been elaborated by an expert group representing Member State Competent Authorities and the European Commission, in consultation with all relevant actors including notified bodies and manufacturers.
In order to promote global convergence, this document takes into account certain concepts outlined in the Global Harmonisation Task Force guidance documents (such as SG5/N7:2012). (1)
2. Scope
This guidance should be applied to all products meeting the definition of an IVD per
Article 2(2) of the IVDR: any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
a) concerning a physiological or pathological process or state;
b) concerning congenital physical or mental impairments;
c) concerning the predisposition to a medical condition or a disease;
d) to determine the safety and compatibility with potential recipients;
e) to predict treatment response or reactions;
f) to define or monitoring therapeutic measures.
Specimen receptacles shall also be deemed to be in vitro diagnostic medical devices;
As accessories for an IVD fall under the scope of the IVDR, this document also provides guidance on these devices.
This document provides guidance on:
– General principles of Clinical Evidence,
– Performance evaluation process,
– The role of risk management in performance evaluation,
– Performance Evaluation Plan (PEP),
– Scientific Validity, Analytical Performance and Clinical Performance,
– Performance Evaluation Report (PER),
– Continuous update of the performance evaluation.
It is important to remind that per Article 1(3) of the IVDR, the following products are not considered IVDs and are henceforth out of scope of this guidance:
a) products for general laboratory use or research-use only products, unless such products, in view of their characteristics, are specifically intended by their manufacturer to be used for in vitro diagnostic examination;
b) invasive sampling products or products which are directly applied to the human body for the purpose of obtaining a specimen;
c) internationally certified reference materials;
d) materials used for external quality assessment scheme.
Please note that this guidance does not elaborate on performance studies in detail nor does it address the concept of equivalence in detail. In addition, this guidance does not apply to in-house devices.
3. Introduction
Prior to placing an IVD on the market or putting it into service, the manufacturer must demonstrate compliance with all applicable requirements of the IVDR, in accordance with the appropriate conformity assessment procedure(s). Therefore, the manufacturer must demonstrate that the IVD achieves its intended purpose in accordance with the claimed performance over the lifetime of the device. (2)
Article 56 (1) of the IVDR underlines that the manufacturer must specify and justify the level of clinical evidence in view of the characteristics of the device and its intended purpose. As such, defining the intended purpose of an IVD must be considered a key driver behind the overall assessment. Accordingly, the full intended purpose should be clearly expressed in the intended purpose statement, reflected in the instructions for use (IFU), and should include the specific elements outlined in Annex I, 20.4.1 (c.):
(i) what is detected and/or measured;
(ii) its function (e.g. screening, monitoring, diagnosis or aid to diagnosis, prognosis, prediction, companion diagnostic);
(iii) the specific information that is intended to be provided in the context of:
— a physiological or pathological state;
— congenital physical or mental impairments;
— the predisposition to a medical condition or a disease;
— the determination of the safety and compatibility with potential recipients;
— the prediction of treatment response or reactions;
— the definition or monitoring of therapeutic measures;
(iv) whether it is automated or not;
(v) whether it is qualitative, semi-quantitative or quantitative;
(vi) the type of specimen(s) required;
(vii) where applicable, the testing population; and
(viii) for companion diagnostics, the International Non-proprietary Name (INN) of the associated medicinal product for which it is a companion test.
In this context, it is relevant to distinguish analytes with specific indications (e.g. specific diagnostic purposes) from those which may be relevant for multiple clinical conditions and are consequently intended to assess physiological status rather than a specific diagnostic indication. In such cases, the intended purpose should be framed to appropriately reflect the IVD’s overall purpose, e.g. ‘as a marker of inflammation’ rather than specifying specific causes of inflammation (unless specifically diagnostic for these). Additional information regarding different clinical contexts for the analyte should be captured in the scientific validity of the IVD and be included in other sections of the IFU, e.g. the ‘summary and explanation of the test’ section.
The IVDR outlines that evidence for an IVD’s conformity is established by demonstrating and substantiating the scientific validity, analytical performance and clinical performance. Furthermore, the IVDR underlines that the necessary clinical evidence should be based on a sufficient amount and quality of data in order to allow a qualified assessment of whether the IVD is safe, performant and achieves the intended clinical benefit(s), when used as intended. The IVDR notes that clinical evidence may include data from devices which are claimed to be equivalent to the device under assessment. The handling of equivalence should be defined in the manufacturers QMS (Annex IX 2.2.c). Where data from equivalent devices is used, a justification should be provided. It is important to underline that risk classification is not the only factor which influences the level of clinical evidence needed. As a general principle, scientifically substantiated conclusions should be reached through the use of a systematic and explicit appraisal of data that supports decisions made and conclusions reached regarding sufficient clinical evidence.
To assist in demonstrating conformity with the IVDR, IVD manufacturers may make use of harmonised standards, or the relevant parts of those standards, the references of which are published in the Official Journal of the European Union. While the use of harmonised standards is recommended, it is not the only means to demonstrate an IVD’s conformity with the IVDR. When other approaches are employed, the manufacturer must ensure that the necessary level of safety and performance is achieved.
Where no harmonised standards exist or where the existing harmonised standards are not sufficient, the Commission can adopt Common Specifications (CS) regarding the requirements on performance evaluation and performance studies. IVDs developed in compliance with CSs (fully or partially), are in presumption of conformity with the requirements of the IVDR covered by the CS or parts thereof. Thereby, manufacturers or study sponsors must comply with the CS unless it can be duly justified that the adoption of a different solution ensures an equivalent level of safety and performance.
It is important to underline that performance evaluation should be regarded as a continuous process required not only to generate but also maintain the clinical evidence needed to support an IVD’s intended purpose. It is with this essence the IVDR requires that a lifecycle approach must be employed, whereby clinical evidence is updated throughout the IVD’s entire lifecycle. Scientific developments and improvements in state-of-the-art should be reviewed and assessed by the manufacturer on a regular basis as part of their continuous and pro-active post- market surveillance activities. Therefore, manufacturers must instate a procedure for planned monitoring of scientific developments and changes in medical practice relevant to the IVD(s). Any relevant new information, developments and progress in the scientific field should trigger reassessments of the existing clinical evidence thus ensuring safety and performance through a continuous performance evaluation process.
4. Definitions
The definitions elaborated within this section and utilised within this document are intended to apply to IVDs according to the IVDR. A small number of terms which are considered useful and defined below were taken from other sources than the IVDR.
Analytical performance
Analytical sensitivity
Analytical specificity
Certified Reference
Material/ Standard
Reference Material
Certified reference methods
Clinical Benefit
Clinical evidence
Clinical performance
Device for performance study
Diagnostic sensitivity
Diagnostic specificity
False negative
False positive
Usability engineering
Investigator
Likelihood ratio
Linearity
Measuring interval (range)
Negative predictive value
Performance evaluation
Performance of a device
Performance study
Performance study plan
Positive predictive value
Predictive value
Risk
Robustness
Scientific validity of an analyte
Specimen
State-of-the-art
Summary of Safety and Performance (SSP)
Metrological Traceability
True negative
True positive
Usability
WHO International Standard
Use environment
The ability of a device to correctly detect or measure a particular analyte.
Source: EU 2017/746 (IVDR), Article 2 (40)
The capability of the method to distinguish between two close concentrations of the target marker/analyte.
Source: Iteration from several sources
The ability of an assay to measure in a sample a particular target measurand in the presence of for example other analyte/marker, matrix, interfering substances/organisms or cross-reactive species/agents.
Source: Iteration from several sources
Reference material, accompanied by a certificate, one or more whose property values are certified by a procedure which establishes its traceability to an accurate realization of the unit in which the property values are expressed, and for which each certified value is accompanied by an uncertainty at a stated level of confidence.
Source: WHO TGS-8 definition: Quality control for in vitro diagnostic medical devices for WHO prequalification
Measurement method which has been certified to show appropriate trueness and precision for its intended purpose and has been officially defined as reference method by a competent body.
Source: WHO TGS-8 definition: Quality control for in vitro diagnostic medical devices for WHO prequalification
The positive impact of a device related to its function, such as that of screening, monitoring, diagnosis or aid to diagnosis of patients, or a positive impact on patient management or public health.
Note: It should be recognized that the concept of clinical benefit for in vitro diagnostic medical devices is fundamentally different from that which applies in the case of pharmaceuticals or of therapeutic medical devices, since the benefit of in vitro diagnostic medical devices lies in providing accurate medical information on patients, where appropriate, assessed against medical information obtained through the use of other diagnostic options and technologies, whereas the final clinical outcome for the patient is dependent on further diagnostic and/or therapeutic options which could be available.
Source: EU 2017/746 (IVDR), Article 2 (37) and recital 64
Clinical data and performance evaluation results pertaining to a device of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s), when used as intended by the manufacturer.
Source: EU 2017/746 (IVDR), Article 2 (36)
The ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user.
Source: EU 2017/746 (IVDR), Article 2 (41)
A device intended by the manufacturer to be used in a performance study.
A device intended to be used for research purposes, without any medical objective, shall not be deemed to be a device for performance study.
Source: EU 2017/746 (IVDR), Article 2 (45)
The ability of a device to identify the presence of a target marker associated with a particular disease or condition.
Source: EU 2017/746 (IVDR), Article 2 (50)
The ability of a device to recognise the absence of a target marker associated with a particular disease or condition.
Source: EU 2017/746 (IVDR), Article 2 (49)
A result where the device incorrectly indicates that the specimen tested negative for the condition, attribute or property under analysis.
A result where the device incorrectly indicates that the specimen tested positive for the condition, attribute or property under analysis.
Application of knowledge about human behaviour, abilities, limitations, and other characteristics to the design of and interactions with an IVD medical devices (including software) to achieve adequate usability.
Source: Modified from IEC 62366
An individual responsible for the conduct of a performance study at a performance study site.
Source: EU 2017/746 (IVDR), Article 2 (48)
Likelihood of a given result arising in an individual with the target clinical condition or physiological state compared to the likelihood of the same result arising in an individual without that clinical condition or physiological state.
Source: EU 2017/746 (IVDR), Article 2 (54)
The ability to provide measured quantity values that are directly proportional to the value of the measurand in the sample.
Source: ISO 18113-1
A set of values of quantities of the same kind that can be measured by a given measuring instrument or measuring system with specified instrumental measurement uncertainty, under defined conditions
Source: International vocabulary of metrology – Basic and general concepts and associated terms (VIM) 3rd edition
The ability of a device to separate true negative results from false negative results for a given attribute in a given population.
Source: EU 2017/746 (IVDR), Article 2 (53)
An assessment and analysis of data to establish or verify the scientific validity, the analytical and, where applicable, the clinical performance of a device.
Source: EU 2017/746 (IVDR), Article 2 (44)
Performance of a device means the ability of a device to achieve its intended purpose as claimed by the manufacturer. It consists of the analytical and, where applicable, the clinical performance supporting that intended purpose.
Source: EU 2017/746 (IVDR), Article 2 (39)
A study undertaken to establish or confirm the analytical or clinical performance of a device.
Source: EU 2017/746 (IVDR), Article 2 (42)
A document that describes the rationale, objectives, design methodology, monitoring, statistical considerations, organisation and conduct of a performance study.
Source: EU 2017/746 (IVDR), Article 2 (43)
The ability of a device to separate true positive results from false positive results for a given attribute in a given population.
Source: EU 2017/746 (IVDR), Article 2 (52)
The probability that a person with a positive device test result has a given condition under investigation, or that a person with a negative device test result does not have a given condition.
Source: EU 2017/746 (IVDR), Article 2 (51)
The combination of the probability of occurrence of harm and the severity of that harm.
Source: Article 2(16) IVDR
The robustness of an analytical procedure means the capacity of an analytical procedure to remain unaffected by small but deliberate variations in method parameters and provides an indication of its reliability during normal usage.
Source: ICH Q2(R1)– Validation of analytical procedures
The association of an analyte with a clinical condition or a physiological state.
Source: EU 2017/746 (IVDR), Article 2 (38)
Is a discrete portion of a body fluid or tissue taken from an individual for examination, study or analysis of one or more quantities or characteristics to determine the character of the whole. This also includes other materials, for example, hair, nails excretions, secretions, or a sample from the skin surface.
Source: MDCG IVD Classification guidance
Developed stage of current technical capability and/or accepted clinical practice in regard to products, processes and patient management, based on the relevant consolidated findings of science, technology and experience.
Note: The state-of-the-art embodies what is currently and generally accepted as good practice in technology and medicine. The state-of-the-art does not necessarily imply the most technologically advanced solution. The state-of-the-art described here is sometimes referred to as the “generally acknowledged state-of-the-art”
Source: Modified from IMDRF/GRRP WG/N47 FINAL:2018
Article 29 specifies the information, including data on performance evaluation and respective conclusions, which manufacturers have to provide to notified bodies for validation and to make available to the public in the “Summary of safety and performance” in the EUDAMED database.
Source: EU 2017/746 (IVDR), Article 29
Property of a measurement result whereby the result can be related to a reference through a documented unbroken chain of calibrations, each contributing to the measurement uncertainty. The metrological traceability chain is a sequence of measurement standards and calibrations that is used to relate a measurement result to a reference.
Source: International vocabulary of metrology – Basic and general concepts and associated terms (VIM) 3rd edition
A result where the device correctly indicates that the specimen tested negative for the condition, attribute or property under analysis.
A result where the device correctly indicates that the specimen tested positive for the condition, attribute or property under analysis.
Characteristic of the user interface that facilitates use and thereby establishes effectiveness, efficiency and user satisfaction in the intended use environment.
Note 1 to entry: all aspects of usability, including effectiveness, efficiency and user satisfaction, can either increase or decrease safety.
Source: IEC 62366:2015
WHO provided international standard for the calibration and validation of diagnostic and screening assays.
Actual conditions and setting in which users interact with the IVDs (e.g. self-tests).
Source: modified from 62366-1:2015
5. General principles of Clinical Evidence
Clinical evidence for IVDs is established through the collection of data as a result of a performance evaluation. Performance evaluation covers the assessment and analysis of data to establish and verify the scientific validity, analytical performance and, where applicable, clinical performance of an IVD as per article 2 (44). Clinical expertise and judgment is required at every step of the performance evaluation, including the collection and appraisal of data arising from different sources. Each indication and claimed clinical benefit specified in the intended purpose should be assessed and have the appropriate supporting clinical evidence.
As defined in Article 2 (37) of the IVDR, clinical benefit is the positive impact of a device related to its function, such as that of screening, monitoring, diagnosis or aid to diagnosis of patients, or a positive impact on patient management or public health. Preamble (64) of the IVDR further clarifies that the concept of clinical benefit for IVDs is fundamentally different from that which applies in the case of pharmaceuticals or of therapeutic medical devices, since the benefit of IVDs lies in providing accurate medical information on patients, where appropriate, assessed against medical information obtained through the use of other diagnostic options and technologies, whereas the final clinical outcome for the patient is dependent on further diagnostic and/or therapeutic options which could be available.
As a substantial percentage of healthcare decisions rely on information provided by IVDs, results from IVDs can significantly influence patient diagnosis, management, treatment and overall clinical outcomes. As such, the clinical evidence of an IVD should demonstrate that the defined clinical benefit is achieved and that the IVD is safe and in conformity with the applicable general safety and performance requirements (GSPRs) set out in Annex I. The clinical evidence must also support the intended purpose and performance of the IVD, as stated by the manufacturer, while addressing the residual risks to the patients, users or other persons associated with the use of the device. As accessories fall under the scope of the IVDR, establishment of the clinical evidence for an accessory used together with one or several IVD(s) may be performed alongside the corresponding IVD(s) in question. To determine and justify the level of clinical evidence, the amount and quality of supporting data should be evaluated. According to the principles of evidence-based medicine, the body of evidence should be assessed taking into the account strength, robustness, and quality of data in order to draw meaningful conclusions. All in all, the clinical benefit of the IVD should always outweigh the overall residual risk.
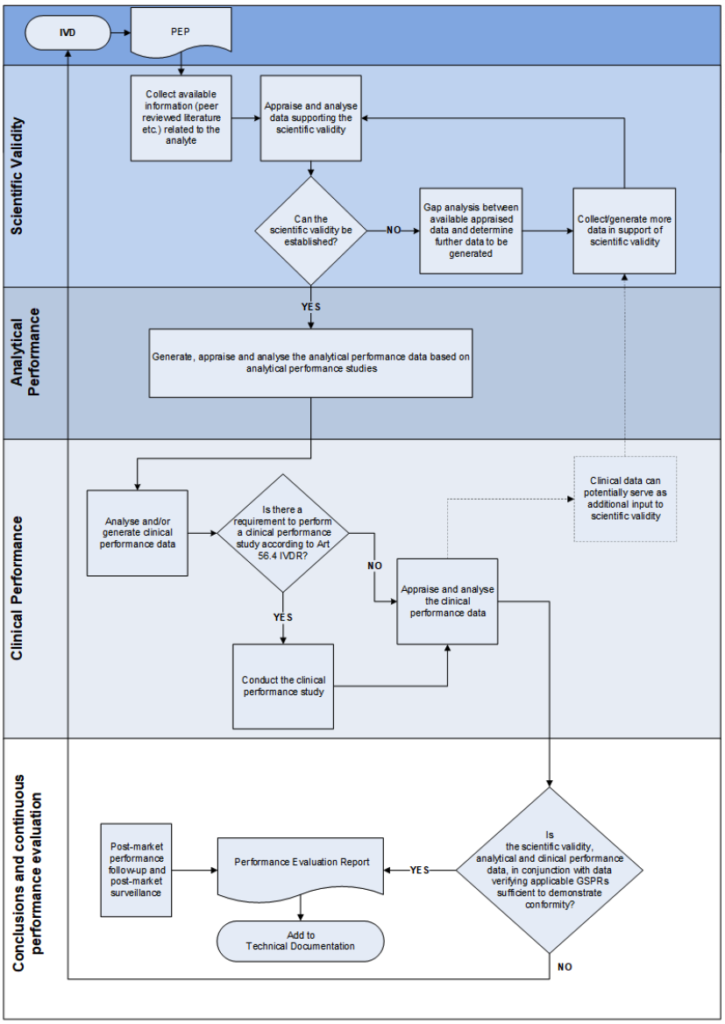
As part of the ‘lifecycle approach’ of a device, it is of utmost importance that the generation and assessment of clinical evidence remains a continuous process during the life-time of the device. The general methodological principles to be considered by the manufacturer are listed in Annex XIII 1.2. of the IVDR and are further illustrated in Appendix I of this guidance document.
6. Performance Evaluation
6.1. Performance evaluation process
Performance Evaluation is a structured, transparent, iterative and continuous process which is part of the quality management system and is conducted throughout the life cycle of an IVD. The general performance evaluation principles are laid down in Article 56 and Annex XIII, Part A, 1 of the IVDR and can be summarised as follows:
1. Planning: establishment and maintenance of a performance evaluation plan (PEP); identification of the approach and steps to generate the necessary clinical evidence based on the characteristics of the device, its intended purpose, etc.;
2. Data Establishment:
– Identification and evaluation of the available data in terms of its suitability and relevance for demonstrating conformity with the relevant GSPRs and intended purpose;
– Identification of whether additional scientific validity, analytical performance or clinical performance data is required to demonstrate conformity and identification of any unaddressed issues or gaps exist in the data;
– Generation of scientific validity, analytical performance and clinical performance data needed (e.g. to address gaps);
3. Analysis, conclusions and documentation: analysis and documentation of the scientific validity, analytical performance data and clinical performance data. Assessments and drawing of conclusions in the performance evaluation report (PER); drawing up of the summary of safety and performance (for Class C and D). (3)
4. Continuous monitoring and updates: Update the performance evaluation report, the summary of safety and performance (for Class C and D) and other associated documentation (e.g. Periodic Safety Update Report (PSUR)) throughout the life cycle of the IVD considering also data obtained from implementation of the manufacturer’s Post Market Performance Follow-up (PMPF) report and through a continuous evaluation of the state-of-the-art.
This approach is illustrated in Figure 1.
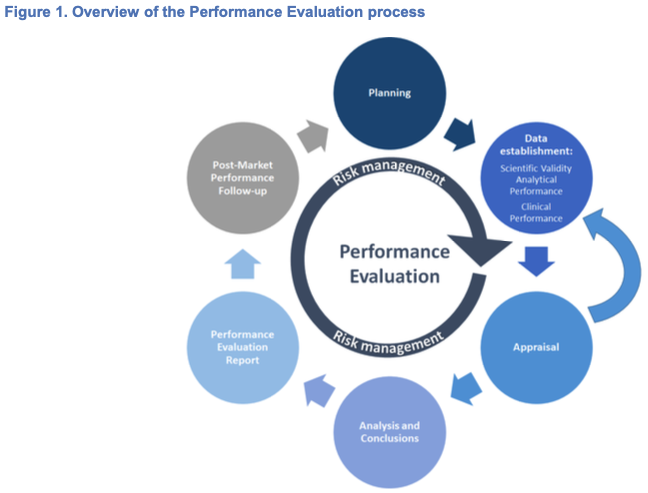
The performance evaluation of an IVD must consider the benefit-risk ratio in light of the state-of-the-art. The three essential pillars of performance evaluation can be summarised as:
– Scientific validity: the extent to which the analyte, or marker to be determined by the IVD is associated with the targeted physiological state or clinical condition. (4)
– Analytical performance: demonstration of the IVD’s ability to correctly detect or measure a particular analyte. (5)
– Clinical performance: demonstration of an IVD’s ability to yield results that are correlated with a particular clinical condition or a physiological/pathological process or state in accordance with the target population and intended user.
When planning the performance evaluation, the approach to demonstrating compliance with the applicable requirements and methods can vary. For devices where CS have been published, aspects of the clinical evidence requirements are clearly defined. Some devices may have relevant existing standards, guidelines (e.g. WHO international standards, best practice documents) and/or certified reference materials or reference measurement procedures. The availability of these may assist manufacturers when designing the PEP (see section 6.3) and can be utilised in studies required to generate sufficient clinical evidence to demonstrate conformity.
For devices that do not typically have, for example, CS or certified reference materials, planning activities to generate necessary clinical evidence may be more challenging. In such cases manufacturers are reminded that the clinical evidence must be sufficient to support the intended purpose of the device in the intended use environment. If there are no comparative methods, different approaches may be used if demonstrated to be appropriate, such as comparison to some other well- documented methods or the composite reference standard. The depth and extent of clinical evidence should also take into account considerations including the inherent risks of the device and the state-of-the-art. It may be worth reflecting on the concepts listed in section 6.8 when designing the PEP and any studies required to generate sufficient clinical evidence.
Devices with, for example, new intended purposes, new analytes, new target populations, or are based on new technologies or technical processes may not have a large amount of existing evidence available due to their novelty. The availability of existing evidence and current clinical practices to support the novel aspects should be a key consideration when designing the PEP and any studies necessary to generate sufficient clinical evidence.
6.2. The role of risk management in performance evaluation
The risk management system should be carefully aligned with and reflected in the performance evaluation process of the IVD, considering the clinical risks to be addressed as part of the performance evaluation, performance studies, and post- market performance follow-up(s).
Due to their nature, in the majority of cases, deficiencies of IVDs do not directly lead to physical injury or damage to the health of people. If any, these devices may lead to indirect harm, rather than direct harm.
The requirements to remove or reduce risks as far as possible must be fulfilled, taking into account the generally acknowledged state-of-the-art in the field of medicine. Risks for IVDs are often generated from a series of events which may involve several factors such as inadequate design characteristics, immature technology or usability, defects in manufacturing, unintentional misuse, improper storage or maintenance of the IVD as well as medical decisions of the healthcare professional or lay user based on the examination results.
Chain of events may lead to hazardous situations, e.g. reporting of an incorrect examination result. As a consequence of a medical decision based on the result provided by the device, action taken or not taken may result in the worst cases to severe harms to the patients. In addition to the harm to patients, exposures of the users (e.g. maintenance personnel, patients) to chemical, electronic, electromagnetic, mechanical (non-exhaustive) should be accounted for.
Therefore, the risk analysis should always reflect the clinical use and medical purpose of the IVD. For more examples on series of events leading to harm, see Table 1.
Table 1. Examples of series of events leading to harm. The probability of occurrence of harm can be combination of probability of the hazardous situation occurring and probability of hazardous situation leading to the harm. Severity of harm is often dependent of intended purpose of the device.
Probability of the Occurrence of the Harm x Severity | ||||
|---|---|---|---|---|
Probability of the Hazardous situation | Probability of the Harm | |||
Manufacturer risk control measures | Clinical risk control measures | |||
An event/A defect | Hazard | Hazardous situation | Clinical events contributing or mitigating the hazardous situation | Harm and Severity |
Poor sampling, stability of the reagent degraded etc. | False negative result/Too low result/Wrong result | Healthcare professional/layman receives an incorrect result | Asymptomatic The result (not) considered in the context of clinical signs and symptoms Not identified patients likely to benefit from the corresponding medicinal product Result accepted as correct Treated/Untreated | Death |
Life-threatening illness or injury | ||||
Hospitalization or prolongation of hospitalization | ||||
Inappropriate medical treatment | ||||
Minor harm not leading to additional medical treatment or treatment, minor inconvenience | ||||
Cross reaction, too high amount of sample dispensed etc. | False positive result/Too high result/Wrong result | Healthcare professional/layperson receives an incorrect result | Asymptomatic The result (not) considered in the context of clinical signs and symptoms Not identified patients likely to be at increased risk of serious adverse reactions as a result of treatment with the corresponding medicinal product Result accepted as correct Treated/Untreated | Death |
Life-threatening illness or injury | ||||
Hospitalization or prolongation of hospitalization | ||||
Inappropriate medical treatment | ||||
Minor harm not leading to additional medical treatment or treatment, minor inconvenience | ||||
Degradation of quality control(s), result cannot be accepted etc. | Delayed result | Healthcare professional do not receive a result in a timely manner | Emergency use, No Delay of medical intervention | Death |
Life-threatening illness or injury | ||||
Hospitalization or prolongation of hospitalization | ||||
Inappropriate medical treatment | ||||
Minor harm not leading to additional medical treatment or treatment, minor inconvenience | ||||
Barcode reading error, a component missing from the kit etc. | No result | Healthcare professional/layperson do not receive a result at all | Emergency use, No Asymptomatic Delay of medical intervention | Death |
Life-threatening illness or injury | ||||
Hospitalization or prolongation of hospitalization | ||||
Inappropriate medical treatment, | ||||
Minor harm not leading to additional medical treatment or treatment, minor inconvenience | ||||
Test line colour/bacteria l growth result interpretation unclear etc. | Unclear result | Healthcare professional/layperson receives an unclear result/do not receive a result in a timely manner | Asymptomatic The result (not) considered in the context of clinical signs and symptoms Result accepted as correct Treated/Untreated | Inappropriate medical treatment, |
Minor harm not leading to additional medical treatment or treatment, minor inconvenience | ||||
Mechanical, Electronic, Electromagnetic, Chemical defect etc. | Electric shock, pain, blood scratch, eye damage, inconvenience | Healthcare professional/ | Death | |
Life-threatening illness or injury | ||||
Hospitalization or prolongation of hospitalization | ||||
Inappropriate medical treatment, | ||||
Minor harm not leading to additional medical treatment or treatment, minor inconvenience | ||||
The manufacturer’s risk management system should define a scientific and practical approach to support decision making. All known and foreseeable risks, and any undesirable effects shall be minimised and be acceptable when weighed against the evaluated potential benefits to the patients and/or the user arising from the intended performance of the device during normal conditions of use.
Risks and risk control measures should be reflected in the performance evaluation process for the device in line with an up-to-date lifecycle approach. For example, the impact on the clinical evidence required to support the use of the device should be considered when new risks are identified or when changes are implemented.
6.3. Performance Evaluation Plan
As highlighted above, the performance evaluation begins with the establishment of the PEP. To generate the necessary clinical evidence, the PEP should specify certain characteristics, performance of the device, the process and the criteria to be applied.
When designing the PEP, manufacturers should take into account the following factors (non-exhaustive) in view of the safety and performance requirements for the device:
– the intended purpose (including for example the specific disorder, condition or risk factor of interest that it is intended to detect, define or differentiate),
– the novelty and degree of innovation,
– the scientific validity of the analyte,
– the specification of methods,
– the assay technology,
– the state-of-the-art,
– the risk to the patient (e.g. due to an incorrect or delayed result from an IVD),
– the intended user(s),
– the disease state(s),
– the device classification,
– the degree of variability of the study subject population,
– prevalence of the clinical state,
– the availability of certified reference materials or certified reference methods,
– the target population,
– the stability of specimens, reagents, etc.
– availability of CS.
The requirements for the PEP are set out in the IVDR Annex XIII.
The manufacturer must plan, continuously conduct and document a performance evaluation and should establish and update the performance evaluation plan (per IVDR Annex XIII Part A 1), and the performance evaluation report with data obtained from the manufacturer’s PMPF (per IVDR Annex XIII Part B 6). Identification and specification of the applicable GSPRs should be clearly outlined within the PEP.
Additionally, an assessment of the benefit-risk ratio should be included in the PEP. To determine the benefit-risk acceptability, pre-determined thresholds indicating and specifying parameters and criteria should be used. This is required in order to determine if the clinical benefits of the intended purpose outweigh the overall residual risk.
Whilst they are neither analytical performance studies nor clinical performance studies, testing conducted in order to prove compliance with the applicable GSPRs (Annex I Chapter II point 10 onwards e.g. electromagnetic compatibility testing or electrical safety testing), would need to be taken into account within the performance evaluation and their results should be reflected in the performance evaluation report.
Figure 2 (below) illustrates the flow of plans and reports within a performance evaluation and describes the relevant information that is required in the development process of an IVD.

Appendix II of this guidance provides an overview of the required frequency of updates for the different documents depending on the device class.
6.4. Scientific Validity
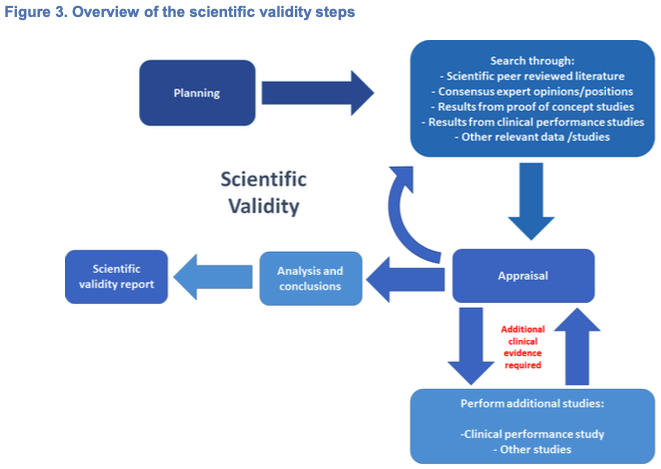
Scientific validity should be demonstrated and documented for each device. As defined by IVDR Article 2(38), the ‘scientific validity of an analyte’ is the association of an analyte with a clinical condition or a physiological state. In the case of IVD MDSW, scientific validity should be demonstrated and documented for each device that provides information on and with respect to a clinical condition or a physiological state. It is important to underline that for certain devices intended to be used together, for example a reagent intended to be used with calibrators and controls, it may be more appropriate to establish the scientific validity in the context of this combination. As certain instruments may have an independent measuring function which does not use any additional reagents, (6) scientific validity would also be required unless due justification is given. Scientific validity may be demonstrated through the use of existing data, where available, while taking into account the generally acknowledged state-of-the-art. If the association of the analyte to a clinical condition or physiological state is well established, evidence may be available in sufficient quality and quantity to substantiate the scientific validity. In such cases, this association may be appropriately demonstrated based on available information such as peer reviewed literature, textbooks, historical data and experience. Where existing evidence does not exist, is considered insufficient, or where there is a high degree of novelty (such as new analytes and/or new intended purposes), it will be necessary to provide a scientific rationale and to generate new or additional data. A gap analysis should be conducted by the manufacturer in order to determine the additional evidence required. For example, this may prompt the need to demonstrate scientific validity through studies such as clinical performance studies.
When conducting literature reviews or other data retrieval methods, manufacturers should employ a systematic approach to identify relevant available data:
1. Define a search protocol (7) before commencing data retrieval. The searching strategy must be thorough and objective, i.e. it should identify all relevant favourable and unfavourable data.
2. Conduct several searches with consolidated and relevant criteria or focus in order to obtain all relevant and necessary data.
3. Ensure appropriate documentation is produced such that the methods can be appraised critically, the results can be verified, and searches reproduced if necessary.
Examples of existing data (without any particular order):
– appraised literature data,
– peer-reviewed data
– published clinical data (e.g. Summary of Safety and Performance (SSP), Registries and databases from authorities),
– relevant information on the scientific validity of devices measuring the same analyte or marker,
– proof of concept studies (8),
– relevant consensus expert opinions/positions from relevant professional associations relating to the safety, performance, clinical benefits to patients, design characteristics, scientific validity, clinical performance and intended purpose of the device.
Examples of generating new evidence (without any particular order)
– Perform clinical performance study,
– Other studies (e.g. analytical performance studies or PMPF studies).
After finalising the collection and/or generation of evidence supporting the scientific validity, the manufacturer should appraise and analyse the obtained data and reflect it within the scientific validity report.
Figure 3 below highlights the different steps the manufacturer may take in order to meet the requirements for establishing scientific validity.
6.5. Analytical Performance
Per IVDR Article 2(40), the analytical performance focuses on the gathering of evidence that the IVD in question reliably, accurately and consistently measures and/or detects an analyte(s).
The relevant performance characteristics, as part of the GSPRs and linked to the analytical features of the IVD should be supported by existing evidence or by generating new evidence. As a general rule, the analytical performance should always be demonstrated on the basis of analytical performance studies. Data from published experience gained by routine diagnostic testing may be considered as supportive evidence to the analytical performance.
The manufacturer should verify that all different specimen types and specimen sampling conditions that are indicated in the IVD’s intended purpose are assessed and demonstrated. (9) To ensure stability, this examination should also include, where applicable, the required devices for specimen collection, the indicated specimen storage and transport conditions.
Information on the timeframe between the specimen collection, storage conditions and its analysis should be provided and reflected in the analytical performance report. This includes but is not limited to duration between collection, storage and analysis, temperature limits and recommended number of freeze/thaw cycles. This is especially pertinent for IVDs that use methods with time-critical analysis.
Analytical performance indicators are typically considered similar or even identical across IVDs. These indicators may depend on the assay technology and the intended use environment. The importance and weighting of different indicators listed in the IVDR’s Annex I Section 9.1 and Annex II section 6.1 should be considered on a case by case basis, as not all may be applicable. However, all omissions should be clearly outlined and justified.
Examples of analytical performance indicators include:
– analytical sensitivity,
o limit of blank (LoB),
o limit of detection (LoD),
o limit of quantitation (LoQ),
– linearity,
– measuring interval/range: LoQ as the lower limit and linearity as the upper limit,
– analytical specificity,
o testing against interferents and cross-reacting substances in the presence of other substances/agents in specimen,
– accuracy,
a) trueness of measurement,
b) precision,
o intermediate precision,
o repeatability,
o reproducibility,
– carryover and cross contamination,
– instrument comparison,
– cut-off value(s),
– use environment,
– stability.
For IVD MDSW the following characteristics may be taken in to account:
– confidentiality,
– integrity,
– reliability,
– generalisability,
– expected data rate or quality,
– usability engineering.
Identification of gaps during the analytical performance assessment could trigger the need for the generation of new or additional evidence. In the case of software this may include, for example, to demonstrate generalisability of a software with real-life datasets.
6.6. Clinical Performance
As per Article 2 (41) of the IVDR, clinical performance means the ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user.
The clinical performance aims to demonstrate that the IVD can achieve clinically relevant outputs through predictable and reliable use by the intended user(s). The manufacturer should demonstrate that the IVD has been tested for the intended use(s), target population(s), use condition(s), operating- and use environment(s) and with all the intended user group(s). Indicators of clinical performance vary and depend strongly on the intended purpose and performance claims.
The IVDR sets out that clinical performance may not be required for certain devices in Article 2 (39). For example, clinical performance data may not be expected for non-sterile specimen receptacles, microscopy glass slides, or some general reagents. In such cases and where due justification is given, a clinical performance report would not be expected. Nevertheless, the remaining aspects of the performance evaluation report including other elements of clinical evidence would still be required unless due justification is given.
For markers that are not specific to a particular condition but are adequately defined by scientific validity to be relevant in multiple clinical settings, separate clinical studies for each clinical setting/indication would not be expected.
For those devices demonstrating clinical performance, the following principles are highlighted as potential sources of clinical performance data:
– data from scientific peer-reviewed literature,
– data from published experience gained by routine diagnostic testing,
– data from clinical performance studies,
– other sources of clinical performance data.
Clinical performance can be characterised by the demonstration and evaluation of applicable aspects of clinical performance for the device in question, such as (non- exhaustive):
– diagnostic sensitivity,
– diagnostic specificity,
– positive predictive value,
– negative predictive value,
– number needed to treat/diagnose (average number of patients that need to be treated/diagnosed in order to have an impact on one person),
– number needed to harm/misdiagnose (number of patients that need to be diagnosed/ treated in order have an adverse effect on one patient),
– positive likelihood ratio,
– negative likelihood ratio,
– odds ratio,
– usability /user interface.
Other parameters may be determined by the manufacturer to be applicable when demonstrating the clinical performance characteristics of the IVD in the intended use environment(s) and may be included in the clinical performance report.
It is important that aspects of clinical performance are assessed in terms of their statistical relevance, e.g. inclusion of confidence interval(s) and interpretation of the impact on robustness of the result with regards to the intended purpose. Where due to specific device characteristics demonstration of conformity with GSPRs based on clinical data is not deemed appropriate, a performance evaluation is still required and a justification shall be provided and documented in the PEP and the corresponding PER.
6.7. Clinical performance studies
When determining what data is needed to demonstrate the safety and performance of IVDs, it is important to consider available existing data and how to bridge any deficits. In the event that data is not available in either sufficient quality or quantity it will need to be generated.
Clinical performance studies are considered necessary to establish or confirm compliance with the relevant GSPRs as regards an IVD’s clinical performance which cannot be determined by scientific validity, analytical performance studies, literature, previous experience gained by routine diagnostic testing or other performance studies. Clinical performance studies should be conducted in line with well- established international guidance in this field, such as the international standard ISO 20916 on clinical performance studies using specimens from human subjects (10) regardless of the classification of the device, unless due justification is provided in accordance with Annex XIII 1.2.3. For example, the manufacturer could justify that the use of ‘other sources of clinical performance data’ may be appropriate, if this can be supported by either literature and/or data from published experience gained by routine diagnostic testing.
Clinical performance studies should always be designed in a manner which specifies the clinical evidence the study intends to generate whilst accounting for potential risks, considering appropriate ethical requirements and ensuring compliance with all applicable legal and regulatory requirements. (11) The Clinical Performance Study Plan (CPSP) should define the rationale, objectives, design, proposed analysis, methodology, monitoring conduct and record-keeping of the clinical performance study (IVDR Annex XIII 2.3.2). (12)
As clinical performance studies should be tailored to the specified intended population, the manufacturer must assess and justify the use of any samples within their performance study in view of the intended purpose and type of IVD. (13)
Clinical performance studies conducted under the IVDD should be considered as ‘other sources of clinical performance data’ per Annex XIII 1.2.3 as they wouldn’t meet the requirements of Annex XIII 2.3. (14). It should be noted that an assessment of quality and completeness of the data is essential to identify any potential gaps. This data should be supported by either literature and/or data from published experience gained by routine diagnostic testing.
It must be noted additional requirements must be met by the manufacturer for certain performance studies, such as studies which require ‘surgically invasive sample-taking only for the purpose of the performance study’, that are ‘interventional clinical performance studies’ or which ‘involve additional invasive procedures or other risks for the subjects of the studies’. (15)
When clinical performance studies are conducted, the data obtained should be documented in a clinical performance study report (Annex XIII 2.3.3), used in the performance evaluation process and be part of the clinical evidence for the IVD.
6.8. Performance Evaluation Report
The manufacturer should compile evidence, determine the benefit-risk and document the performance evaluation and its output in the PER. The manufacturer should assess all relevant scientific validity, analytical and clinical performance data to verify the applicable conformity of its device with the general safety and performance requirements as referred to in Annex I. Annex XIII, Section 3 makes reference to studies other than clinical performance studies, these other studies could be analytical performance studies or PMPF studies that should be documented by analogy to clinical performance studies and reflected in the PER.
The amount and quality of data collected should allow the manufacturer to make a qualified assessment whether the IVD will achieve the intended clinical benefit(s) and safety, when used as intended by the manufacturer. The data and conclusions drawn from this assessment constitute the clinical evidence and should take into account the following considerations:
– the intended users,
– the state-of-the-art,
– the nature, severity and the evolution of the condition being diagnosed or treated,
– the adequacy of the estimation of associated risk for each identified hazard,
– the number and severity of adverse events,
– the availability of alternative diagnostic devices and current standard of care.
The assessment may be guided by the following non-exhaustive questions:
– Does the data support the intended purpose, intended users indications, device specifications, target groups, clinical claims and the relevant general safety and performance requirements?
– Has the novelty and level of innovation/history on the market been evaluated and considered?
– Have the risks been identified, mitigated and the effectiveness of the risk control measures been verified?
– Have for example environmental conditions, interference factors, exogenous factors and endogenous factors been evaluated?
– Has the quality of literature retrieved and reviewed been evaluated and has a rationale for the selection process been provided?
– Has there been a sufficient number of observations to draw scientifically valid conclusions?
– Have any limitations within the observations been appropriately justified?
– Was the statistical approach including sample size appropriate to reach a scientifically valid conclusion?
– Have the scientific validity, analytical and clinical performances been demonstrated?
– Is data from performance studies or other sources sufficient to verify the safety and performance, including clinical benefits (where applicable) of the device when used as intended with respect to the state-of-the-art?
– Does the design and results of the performance studies support the clinical evidence?
– Have all deviations from and all planned changes to the performance evaluation plan been justified?
– Has the relevance of the information of the performance evaluation been assessed and documented?
– Has the contribution of each data set to the performance evaluation been weighted according to systematic criteria?
– Is the data set appropriate and takes into account the state-of-the-art of the device?
– Is all supporting data fully traceable, documented and is integrity assured?
– Were all ethical, legal and regulatory considerations/ requirements taken into account?
– Have all omissions been clearly outlined and justified?
7. Continuous update of the performance evaluation
As described previously, performance evaluation is a continuous process conducted over the lifecycle of the IVD. The safety, effectiveness and performance of the IVD is maintained by data which is actively and continuously monitored and collected by the manufacturer. Such data may include, developments in the state-of-the-art, post- market information such as complaints, PMPF data, direct end-user feedback or newly published research, guidelines, harmonised standards.
In addition, changes to an IVD’s e.g. intended purpose, product design, characteristics or technology should be evaluated and the clinical evidence of the IVD should be updated or re-established before such changes are implemented. Developments or changes in the state-of-the-art, harmonised standards or CS by reference to which the conformity of an IVD is declared should be taken into account as they may trigger further activities to update the clinical evidence.
This information should be subject to the performance evaluation process depicted in Figure 1.
7.1. Post-Market Surveillance (PMS) and Post-Market Performance Follow up (PMPF)
In the context of continuous update of the performance evaluation, planning of thorough post market surveillance activities are critical to detect signals and ensure continued performance (e.g. monitor for possible shifts that may signify the emergence of mutated strains not detected by a specific assay).
The IVDR sets out that a PMPF is required to confirm that the IVD’s safety, performance, and clinical evidence throughout its expected lifetime is based on factual evidence and that the benefit-risk ratio remains acceptable. In some cases, PMPF can be justified as not required (Annex III 1b and Annex XIII Part B 8). If PMPF is not deemed appropriate for a specific IVD, a justification should be provided and documented within the performance evaluation report.
Post-market information such as data from the manufacturer’s post-market surveillance system (e.g. serious incident reports, results from post-market performance studies) should be reviewed on a regular basis and used to determine the potential impact on the risks, clinical benefit and whether there is a need to update the performance evaluation report of the IVD. A manufacturer must ensure that appropriate methods, procedures and product specific appropriate triggers for proactively collecting and evaluating safety, performance and scientific data with the aim of conducting PMPF are included in the PMPF plan.
The performance evaluation documentation (PEP & PER) should be updated accordingly as part of the PMPF and the clinical evidence for the IVD expanded as appropriate. Both favourable and unfavourable data should be equally considered. Activities and triggers should be identified to prompt a review of the PMPF. These may include for example:
– Monitoring and analysis of data from post-market use,
– Assessment of published experience gained by routine diagnostic testing,
– Involvement in external quality control schemes,
– Identification of new mutations, strains or variants which may impact the performance of the IVD,
– Inputs from post-market surveillance, including information on serious incident reports and field safety corrective actions.
– Post-market performance studies.
Information gained through the review and assessment of PMPF data may be useful to inform future developments of the device e.g. expansion of the intended purpose or change in claims or design to improve effectiveness.
Appendix I – Methodological principle for generation of clinical evidence
Appendix II Required frequency for updates of reports
| Device Class | Document | Required frequency of update | Article |
|---|---|---|---|
| All | Performance Evaluation and associated documentation | Throughout the life cycle of the device. | Article 56(6) |
| All | Post Market Surveillance Plan | As necessary | Article 79 |
| All | Post-Market Performance Follow-Up (PMPF) | Continuously through product’s lifetime based on input from post market surveillance plan (Annex III) As necessary, unless specific justification is given. | Annex XIII |
| A & B | Performance Evaluation Report | Continuously through product’s lifetime based on input from PMS and PMPF for all classes. As necessary and as defined in the PMS plan | Article 56(6) |
| A & B | Post Market Surveillance Report | When necessary and made available to the notified body and the competent authority upon request | Article 80 |
| C & D | Performance Evaluation Report | As necessary and at least annually | Article 56(6) |
| C & D | Periodic Safety Update Report (PSUR) | At least annually | Article 81(1) |
| C & D | Summary of Safety and Performance (SSP) | As soon as possible, where necessary | Article 29 Article 56(6) |
Footnotes
(1): It shall be noted that as of 2012, the International Medical Device Regulators Forum (IMDRF) took up the work and mission of the Global Harmonization Task Force (GHTF).
(2): During the normal stated conditions of use by the intended user in the intended environment (e.g. laboratories, physician’s offices, healthcare centres, and home environments.)
(3): IVDR Art. 29
(4): For IVD medical device software, analyte or marker should be understood as the association of the claimed intended purpose with a clinical condition or physiological state.
(5): In the case of IVD software, and broadly speaking, accurately, reliably and precisely produce the intended output.
(6): See MDCG 2020-16 Rule 5b.
(7): The search protocol is not a separate deliverable but is rather a part of the performance evaluation plan, the results of which are embedded within the relevant reports.
(8): Proof of concept studies are usually smaller scale scientific studies to identify the fundamental association of the analyte with the clinical condition/physiological state (GHTF/SG5/N7/2012). They are typically used in the feasibility stage of an IVD’s development.
(9): Please note that national provisions on informed consent/ethics approval prior to specimen collection and use may be applicable.
(10): Recital 66 IVDR Corrigendum to Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU (Official Journal of the European Union L 117 of 5 May 2017)(L117/11, 3.5.2019)
(11): For example, the IVDR sets out a requirement for data management. This refers to the process of how data will be captured and managed. Where relevant, it would be appropriate to state how the requirements of the General Data Protection Regulation (GDPR) – Regulation (EU) 2016/679 are being met within the data management process.
(12): If applicable, monitoring plans should be established in order to monitor the study conduct and progress, this will ensure integrity of data, and adequate qualification of study conduction personnel.
(13): Please note that national provisions on informed consent/ethics approval prior to specimen collection and use may be applicable.
(14): Unless the requirements of section 2.3 are appropriately justified.
(15): Article 58(1) IVDR