
MDCG 2022-11 Rev.1
MDCG Position Paper
Notice to manufacturers and notified bodies to ensure timely compliance with MDR and IVDR requirements
Disclaimer: This document is an interactive version of the original MDCG document. We will keep it up-to-date.
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.
MDCG 2022-11 Rev.1 changes
| MDCG 2022-11 revision 1 changes | ||
|---|---|---|
| Section “Call to notified bodies to streamline the certification process” | New | |
| Section “Call to manufacturers to transition to the Regulations and submit their applications for certification without further delay” | Revised | |
With the adoption of Regulations (EU) 2017/745 (MDR) and 2017/746 (IVDR), the regulatory framework for medical devices and in vitro diagnostic medical devices (IVD) has significantly changed. The main objectives of these two Regulations are to establish a robust, transparent, predictable and sustainable regulatory framework for medical devices and in vitro diagnostic medical devices which ensures a high level of safety and health whilst supporting innovation.
Six years have passed since the adoption of MDR and IVDR and the implementation of the regulations is advancing. The Regulations’ transitional provisions have been amended (1) to give manufacturers and notified bodies more time to conduct the necessary conformity assessment procedures and to avert shortages of devices needed for the EU healthcare systems.
Enhancing the capacity of notified bodies and the efficiency of conformity assessment procedures remain a top priority for the MDCG. The actions listed in MDCG 2022-14 are showing good results, e.g. use of hybrid audits, deferral of re-assessment of notified bodies and clarification of the meaning of ‘personnel employed by the notified body’ in Article 36 (1) MDR / Article 32 (1) IVDR. The MDCG remains fully committed to pursue the implementation of the actions listed in MDCG 2022-14 and calls for the continued full commitment of other actors involved, including notified bodies and industry.
To date, 40 and 12 notified bodies are designated under the MDR and the IVDR respectively. The efficiency of the designation process has improved, and individual notified bodies have increased their internal capacity to be able to handle more applications for conformity assessment.
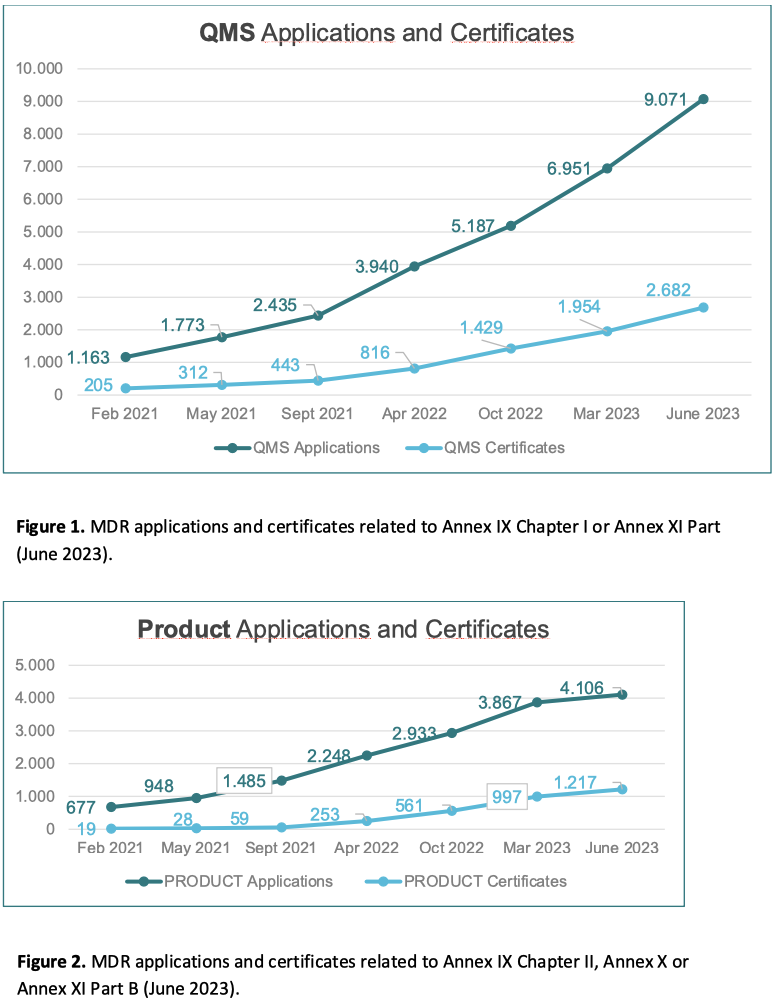
However, data provided by notified bodies2 in June 2023 shows limited progress in terms of applications submitted and certificates issued. Compared to the over 24.000 certificates issued under Directives 90/385/EEC and 93/42/EEC, only around 13.000 MDR applications have been lodged and 3.900 certificates have been issued (see below Figures 1 and 2, page 5). Moreover, out of those 3.900 issued MDR certificates, around 1.000 are related to updates. This shows that manufacturers tend to transfer at different times devices to be included in the same certificate. Whilst this approach is understandable, it might create issues in planning and in the capacity of notified bodies.
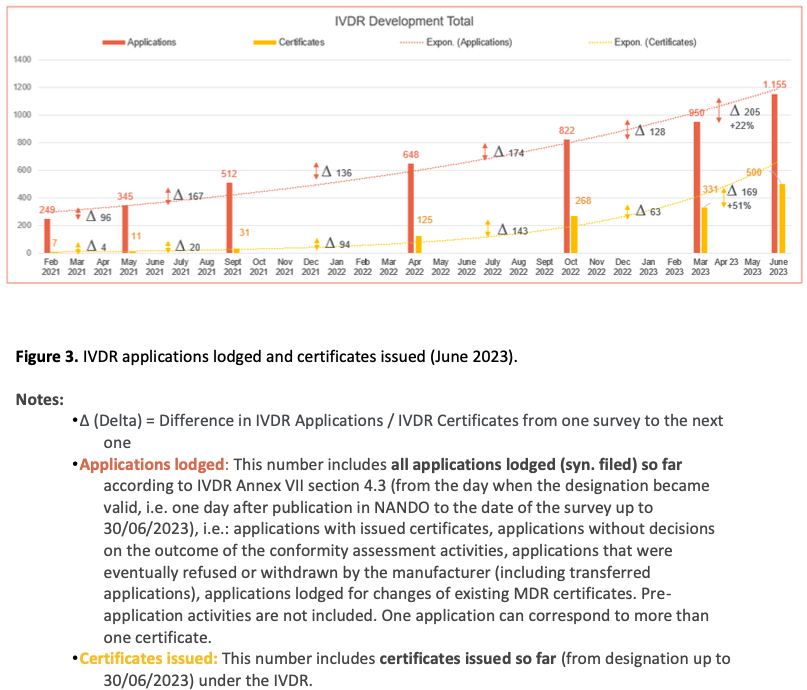
The situation is even more worrisome for IVDs. While only around 8% of IVDs required notified body involvement for conformity assessment under Directive 98/79/EC on in vitro diagnostic medical devices (IVDD), this proportion is around 80% under the IVDR (3). Nonetheless, compared to around 1.500 certificates issued under the IVDD (representing a small fraction of the number of certificates expected under the IVDR), by June 2023 only 1.150 applications have been lodged under the IVDR and around 500 certificates have been issued (see below Figure 3, page 6).
Call to manufacturers to transition to the Regulations and submit their applications for certification without further delay
The MDCG acknowledges the effort of the many manufacturers that already adapted their system to the Regulations. However, the above numbers show that applications submitted by manufacturers, for both Regulations but especially for IVDR, remain low. It appears that manufacturers should continue to strengthen their efforts to transition their products and to initiate the certification process.
Manufacturers should keep in mind that the duration of the conformity assessment process once the application is submitted to a notified body currently takes longer than under the former directives (4). There are several contributing factors to this, including the incompleteness of applications (5). Data from the notified bodies shows that a majority of manufacturers’ applications are incomplete, which may contribute to delays in the certification process.
To improve the efficiency of the process and ensure a smooth transition, the MDCG urges manufacturers to make the best possible use of the additional time provided by the amendments of the MDR and IVDR by submitting applications for conformity assessment in good time.
Considering the deadlines established by the Regulations, manufacturers are thus urged to strengthen their efforts to transition as soon as possible and not to delay submissions further, as this could lead to bottlenecks (6) in the work of notified bodies and possible shortages of products in the market. This is particularly true for manufacturers of class D IVD devices, which must transition to IVDR by May 2025. Notified bodies have strongly recommended applying before the end of 2023 (7). Moreover, it is of utmost importance that the applications to notified bodies are complete and of high quality.
To allow continuous monitoring of the progress of the implementation of MDR and IVDR, the MDCG calls on manufacturers to regularly provide data on the situation regarding their devices. In particular, manufacturers are asked to work closely with authorities and the European Commission to increase transparency and improve the exchange of information (8) on specific medical devices to allow Member States and the European institutions time to prepare for changes to product ranges.
Call to notified bodies to streamline the certification process
The MDCG calls on notified bodies to make the certification process more efficient, transparent and predictable.
It is essential that notified bodies streamline their procedures and make all needed efforts to improve their conformity assessment activities in terms of transparency, timelines, predictability and consistency. Notified bodies are reminded to operate in accordance with consistent, fair, and reasonable terms and conditions, taking into account in particular the interests of SMEs in relation to fees (9).
A proper assistance to manufacturers with regulatory guidance and technical information on how to apply for the conformity assessment procedure (10) is key for a smoother and faster process and to avoid incomplete applications, which have been identified as an important cause of the delays. Such assistance is particularly relevant for SMEs. The MDCG reminds notified bodies to organise structured dialogues with manufacturers (11), which is expected to be part of the normal pre-application and conformity assessment activities and therefore not to be a separated service to be charged for.
To allow continuous monitoring of the progress of the implementation of MDR and IVDR, the MDCG calls on notified bodies to regularly provide data on the situation regarding the certification of devices.
The MDCG calls on notified bodies to increase transparency around their capacity and timelines for conformity assessment and specifically to make publicly available, ideally on a common website, specific information on capacity available at each notified body across the EU for both MDR and IVDR.
Footnotes
(1): Regulation (EU) 2023/607 extending the transitional period according to the risk class of the legacy device until December 2027 or December 2028 and Regulation (EU) 2022/112 extending the transitional period according to the risk class of the legacy device until May 2025 for devices already certified by a notified body under the Directive and class D devices), until May 2026 for class C devices and until May 2027 for class B and A sterile devices).
(2): All data mentioned in this note refers to the Notified Bodies survey, currently managed by Gesundheit Österreich GmbH and Arété on behalf of the European Commission, last update June 2023. https://health.ec.europa.eu/system/files/2023-11/md_nb_survey_certifications_applications_en.pdf.
(3): Preliminary results for 8 – 28 July 2021 Survey Market composition of in vitro diagnostic medical devices (IVDs) EU Competent Authorities for Medical Devices (CAMD) coordinated by MedTech Europe. Link: Analysing the availability of IVDs in May 2022 – MedTech Europe.
(4): Estimation from notified bodies: time for assessment under MDD on average was up to 12 months, under MDR up to 18 or even 24 months; for IVDD on average up to 12 months and under IVDR up to 18 months.
(5): See results of latest survey of Notified Bodies, currently managed via Gesundheit Österreich GmbH, last update June 2023.
(6): To be noted that according to Article 120 (3c) of MDR, manufacturers must lodge an application by 26 May 2024 to benefit from the extended transitional provisions.
(7): To note that the estimated time to complete an IVDR certification process requiring also a product certificate is on average between 13-18 months and 19-24 months and that the end of transitional period for class D devices is May 2025.
(8): E.g. in the context of the study supporting the monitoring of the availability of medical devices on the EU market menages by Gesundheit Österreich GmbH: https://jasmin.goeg.at/2858/1/Study%20supporting%20the%20monitoring%20of%20the%20avaliabiliy_bf.pdf.
(9): Section 1.2.8 of Annex VII to the MDR and to the IVDR.
(10): In accordance with Section 1.2.9. of Annex VII to the MDR and IVDR, the independence and impartiality requirements laid down in Section 1.2. “in no way preclude exchanges of technical information and regulatory guidance between a notified body and a manufacturer applying for conformity assessment.
(11): See: 2022-14 action 15: The MDCG encourages notified bodies and manufacturers to organise structured dialogues before and during the conformity assessment process aimed at regulatory procedures where this is useful to enhance the efficiency and predictability of the conformity assessment process, while respecting the independence and impartiality of the notified body. Such dialogues should not be considered consultancy service.
Revision History
Redline Version
November 2022