
MDCG 2021-6 Rev.1
Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation
Disclaimer: This document is an interactive version of the original MDCG document. We will keep it up-to-date.
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.
MDCG 2020-10/1 Rev.1 changes
| MDCG 2021-6 Rev 1 changes | |
|---|---|
| New questions and answers | Q 2, Q 4, Q 5, Q 14, Q 15, Q 16, Q 17, Q 18, Q 25, Q 26, Q 27, Q 28, Q 39, Q 41, Q 43, Q 44, Q 48, Q 49, Q 50, |
| Updated answers | Q 7 (former Q 4) Q 9 (former Q 6) Q 11 (former Q 7) Q 12 (former Q 8) Q 21 (former Q 11) Q 24 (former Q 14) Q 30 (former Q 15) Q 32 (former Q 17) + Q modified Q 34 (former Q 19) Q 35 (former Q 20) Q 36 (former Q 21) |
| Updated questions | Q 40 (former Q 24) Q 45 (former Q 26) |
| Annexes | Annex I – legend updated Annex II – edits for alignment with MDR terminology Annex III – new |
Table of Contents
Abbreviations
Active Implantable Medical Device Directive, referring to Directive 90/385/EEC
Marking on a product to signify that it meets the legal requirements to be sold on the extended Single Market in the European Economic Area (EEA).
Clinical investigation plan
Clinical Trial Application
Clinical Trial Regulation referring to Regulation (EU) 536/2014 on clinical trials for medicinal products for human use
European Economic Community
European Economic Area
European Norm
European Union
European Database on Medical Devices
General Data Protection Regulation, referring to Regulation (EU) 2016/679 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data
Investigator’s Brochure
The International Organization for Standardization
In Vitro Diagnostic Medical Devices Regulation, referring to Regulation (EU) 2017/746 on in vitro diagnostic medical devices
Medical Device Coordination Group
Medical Device Directive, referring to directive 93/42/EEC
Medical Devices Regulation, referring to Regulation (EU) 2017/745 on medical devices
Member State
National Competent Authority
Post-market clinical follow-up
Serious Adverse Events
Introduction
This document is intended for sponsors of clinical investigations of devices conducted within the scope of the Regulation (EU) 2017/745 (MDR). This document may be supplemented in due course with further questions and answers.
Throughout this document the term ꞌdeviceꞌ is used with the same meaning as in the MDR, i.e., for the purpose of the MDR, medical devices, accessories for medical devices and products listed in Annex XVI of the MDR and to which the MDR applies, shall hereinafter be referred to as ꞌdevicesꞌ (1).
Further, the term “clinical investigation” is used throughout with same meaning as in the MDR Article 2(45), i.e. “any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device”.
While clinical investigation is clearly defined in the MDR, it is sometimes also necessary to mention that there are studies that do not fulfil this definition, to draw the line between when the provisions in the MDR chapter VI and annex XV apply, and when they don’t. For this purpose, the broader term “clinical study” is used, and covers, for the purpose of this guidance, studies done within medical research involving humans and includes clinical trials of medicines, clinical investigations of devices, and clinical performance studies of in vitro diagnostic devices, as well as other studies in a clinical setting where products such as drugs or devices are not necessarily involved at all. (2, 3)
Further, the sponsor (4) needs to be aware that MDR does not specify details about ethics review of clinical investigations. It is thus necessary to check national requirements in relation to submission to the Ethics Committee and if applicable, make sure that Ethics Committees and Competent Authorities have access to the same versions of updated documents.
1. What are the general differences and improvements related to clinical investigations under the new Regulation (EU) 2017/745 (MDR) as compared to the Directives 93/42/EEC and 90/385/EEC?
Regulation (EU) 2017/745 (MDR) will progressively replace both Directives (93/42/EEC and 90/385/EEC) and their transpositions in national law.
The first difference is regarding the type of the law. A Directive is a legislative act that sets out a goal that all EU countries must achieve. However, it is up to the individual countries how to reach these goals by the implementation of national laws. A Regulation, as opposed to a Directive, is a binding legislative act that must be applied in its entirety across the EU on the date of application. It means that the rules are applied in an identical manner throughout the EU. Member States, in authorising and supervising the conduct of a clinical investigation, will be required to base their assessments and decisions on the same rules.
The MDR contains greater detail than the Directive, which is a result of implementing aspects related to good clinical practice, many of which have previously been present in the form of guidance and standard documents.
Further harmonisation at European level will provide greater certainty, which will support an environment that provides greater predictability and is more favourable for conducting clinical investigations, with the highest standards of patient safety, for all EU Member States. It will not only harmonise decisions, but also foster work sharing and collaboration between Member States and enhance the transparency regarding these studies.
For certain clinical investigations, (5) the sponsor still needs to check and follow any specific national provisions which may apply.
2. Is there legislation other than the MDR that needs to be considered when conducting clinical investigations of devices?
The MDR allows, and for some aspects requires, Member States to have national legislation in place, when it comes to e.g. procedures for review and authorisation by ethics committees, responsibility for medical care provided to subjects, investigator qualifications, legally designated representative for subjects, damage compensation systems, designation of competent authority as well as additional requirements for clinical investigations falling under Article 82 of the MDR. For support on applicable national legislation, consult the websites of national competent authorities and ethics committees.
Further, in addition to the MDR there is other European Union legislation that may be applicable, depending on whether other products than devices are being studied in the same clinical study (6), such as the EU Regulation 536/2014 (CTR) (additional information is available under questions 14 and 15 below) and EU Regulation 2017/746 (IVDR) (refer also to Annex III: Does my combination product study require an MDR clinical investigation submission?).
Complying with the MDR does not remove the obligation of sponsors to comply with other relevant legislation, issued by the European Union (7) or Member States, which is not specifically mentioned above.
3. What is a clinical investigation?
A clinical investigation is defined by the MDR as any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device (8).
4. Are all clinical studies that involve use of devices considered clinical investigations as defined by the MDR ??
No, there are situations when devices are used in clinical research but where the scope is not to make a systematic investigation involving one or more human subjects, to assess the safety or performance of the device(s) used. For example, a clinical trial of a medicinal product may involve determining blood pressure or oxygen saturation. This will require the use of devices but since the safety or performance of the devices used for blood pressure/oxygen saturation are not being assessed, such studies would not be considered clinical investigations and devices used in these studies are not investigational devices as defined by the MDR (9).
Note that in these situations, the devices in question will have to be placed on the market or put into service in a manner which is compliant with the MDR (10). Other national and EU legislation (11) regarding aspects such as clinical research, ethical review and personal data protection may also be applicable.
Refer also to question 19 below.
5. Are all clinical investigations of devices covered by the MDR requirements?
The MDR includes requirements for those clinical investigations that are intended to gather clinical evidence for the purpose of demonstrating conformity of devices, and also basic requirements regarding other types of clinical investigations.
Clinical investigations that are carried out as part of the clinical evaluation for conformity assessment purposes are regulated in Article 62(1) of the MDR and need to be designed, authorised, conducted, recorded and reported in accordance with the provisions in Articles 62-81 of the MDR. There may be one or more purposes of such clinical investigations, e.g. to establish and/or verify performance, clinical benefits, clinical safety or any undesirable side-effects.
Article 74 of the MDR lists particular requirements for some clinical investigations of CE-marked devices, see also question 11 below.
Other clinical investigations, i.e. those performed for purposes for which neither Article 62(1) nor Article 74(1) applies, fall under Article 82 of the MDR, which lists minimum requirements that apply to all such clinical investigations and allows Member States to define additional national requirements. For such clinical investigation, you have to check and follow the applicable national provisions in each Member State where the clinical investigation will be conducted.
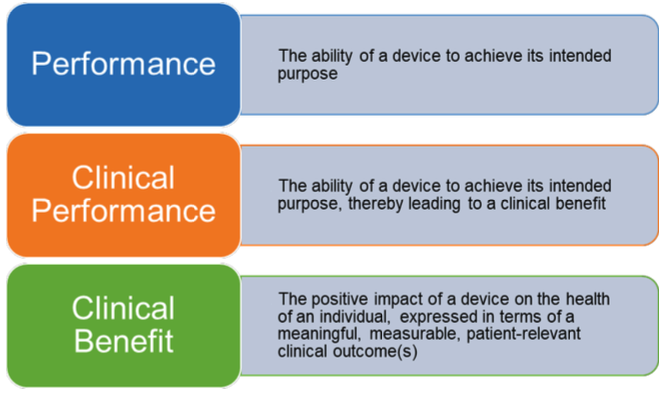
6. What is the difference between the performance, clinical performance and clinical benefit?
In accordance with the MDR, the performance (12) of a device is its ability to achieve its intended purpose as stated by the manufacturer (13). By extension, the clinical performance (14) of a device is the ability of the device to achieve its intended purpose, thereby leading to a clinical benefit (15) when used as intended. Clinical benefit means the positive impact of a device on the health of an individual, expressed in terms of a meaningful, measurable, patient-relevant clinical outcome(s), including outcome(s) related to diagnosis, or a positive impact on patient management or public health.
7. Which regulatory pathway shall a sponsor follow in order to conduct a clinical investigation to collect clinical data that will be used to support the conformity assessment procedure of the investigated device?
Article 62(1) of the MDR provides that the clinical investigations carried out, as part of the clinical evaluation for conformity assessment purposes, shall be designed, authorised, conducted, recorded and reported in accordance with the provisions of Articles 62 to 80 of the MDR. There may be one or more purposes of clinical investigations; e.g. to establish and/or verify performance, clinical benefits, clinical safety and any undesirable side-effects. Such clinical investigations are conducted with devices not yet bearing the CE marking or CE marked devices which are being investigated outside their intended purpose.
For clinical investigations that fall under Article 70(7) point (a) of the MDR (i.e. devices in class I, non-invasive class IIa or class IIb devices), it may be necessary to check national provisions.
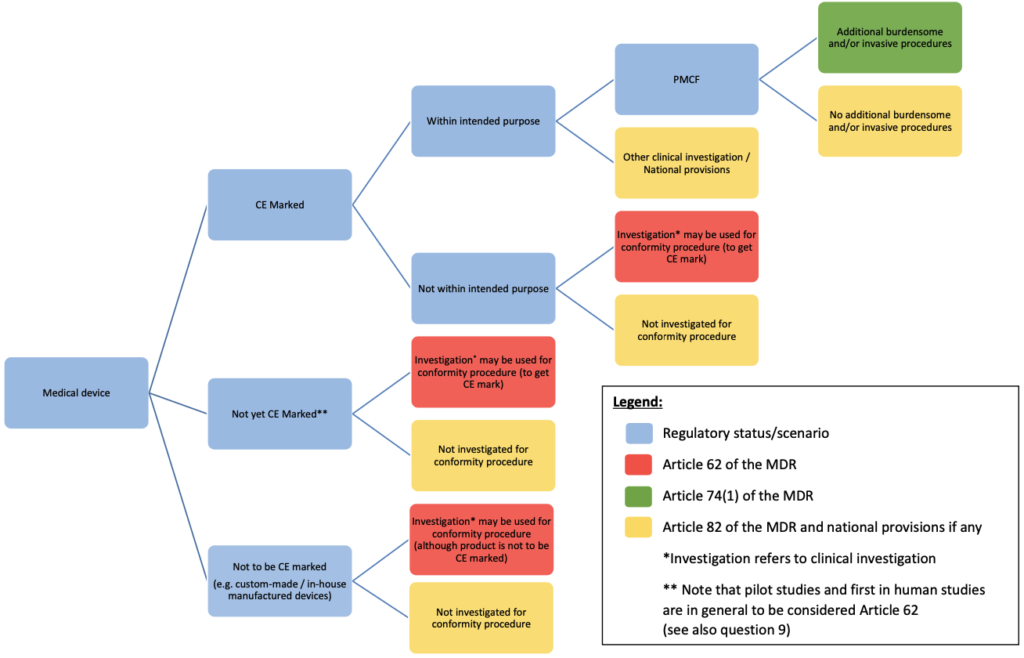
For clinical investigations of other than these devices, the authorisation procedure is defined in Article 70(7) point (b) of the MDR. An overview of the different pathways outlined in the MDR, including national requirements possible under Article 82 of the MDR, is described in Annex I of this document.
8. Which clinical investigations would be considered pilot stage?
A pilot stage clinical investigation is typically an early-stage clinical investigation, which includes the following types:
- First in human clinical investigation
- Early feasibility clinical investigation (may also be called proof-of-concept investigation)
- Traditional feasibility clinical investigation
In general, pilot stage clinical investigations are designed to enrol a limited number of subjects to assess a device early in its development phase with respect to the initial clinical safety and/or performance (e.g. device functionality) (16). The results of this kind of clinical investigation may guide further device design modifications or provide further information for the design of a subsequent clinical investigation. The outcomes of an early-stage clinical investigation can often support further development and iterative changes to the device. The data generated in pilot stage clinical investigations are in general insufficient to CE mark the device but should be considered in the clinical evaluation.
More information on the different development stages and related types of clinical investigation design for investigational medical devices may be found in the standard ISO14155:2020 and its Annex I (17).
9. Which regulatory pathway should a sponsor follow to conduct a clinical investigation in the pilot stage (i.e. first in human, early feasibility) in accordance with the Regulation (EU) 2017/745 (MDR)?
In general, as pilot stage clinical investigations are conducted to gather preliminary safety and/or performance data, the use of Article 62 of the MDR should be foreseen. In cases of doubt, it is recommended to apply under Article 62 of the MDR. Annex I of this document may guide you in the different possible pathways to apply for a clinical investigation.
10. Is it possible to combine several clinical development stages (e.g. pilot/pivotal) in the same clinical investigation?
In some situations, a sponsor may consider it appropriate to include two different study designs in the same CIP (exploratory and confirmatory) to investigate a device for the same intended use and clinical indication as it progresses through clinical developmental stages (e.g. pilot and pivotal stage). This approach should be used restrictively, as the results from the first stage may provide important input to what is the most suitable study design for the subsequent development stage, but there are situations where the approach could be justified.
Note that applications regarding clinical investigations with a phased approach may be subject to conditional approval, where the condition could be that a substantial modification notification to the CAs (including data on outcomes of the first stage) is required before the second stage is initiated.
11. A clinical investigation is planned to further investigate a CE marked device – how does a sponsor determine the regulatory pathway for this clinical investigation?
To determine the regulatory pathway for studies with CE marked devices it is necessary to understand what the intended purpose of the device is and to check whether the planned use in the clinical investigation is within its intended purpose.
The regulatory pathways available for studies with CE marked devices are outlined in Annex I of this document. Question 12 provides further guidance on how to assess the planned use in the clinical investigation with respect to its intended purpose.
Once it has been determined if the investigational medical device will be used within its intended purpose, please refer to the following to determine the appropriate regulatory pathway:
a) When the CE marked device is further assessed, for safety or performance within the intended purpose, this may be done as part of the manufacturer’s post market clinical follow-up activities as a post-market clinical follow-up (PMCF) investigation. Where the PMCF investigation would involve submitting subjects to procedures additional to those performed under the normal conditions of use of the device and those additional procedures are invasive or burdensome, the sponsor shall notify the Member State(s) concerned at least 30 days prior to its commencement, in accordance with Article 74(1) of the MDR. If the sponsor is uncertain whether such additional procedures are considered invasive or burdensome, they are encouraged to request the opinion of the relevant authority in the Member State(s) prior to commencement of the investigation. See question 13 for further information.
b) There are PMCF investigations other than those that need to be notified according to Article 74(1) of the MDR, that do not involve submitting subjects to procedures additional to those performed under the normal conditions of use of the device or where those additional procedures are not considered invasive or burdensome. There are also post market clinical investigations that are not part of the manufacturer’s PMCF activities, such as clinical investigations initiated by other stakeholders and conducted independently from the manufacturer.
If safety and/or performance of a CE marked device is being further investigated, the device will be used within its intended purpose, and if Article 74(1) of the MDR is not applicable, Article 82 of the MDR applies, taking into account national legislation. It is also necessary to check and follow national provisions in the Member State where the clinical investigation will be conducted. Registration in a publicly available database of clinical investigations falling under Article 82 of the MDR is encouraged.
c) When the CE marked device is assessed outside its intended purpose, Article 74(2) of the MDR provides that the requirements for pre-market clinical investigation apply, (Articles 62 to 81 of the MDR). If the clinical investigation is not performed as part of the clinical evaluation for conformity assessment purposes, Article 82 of the MDR applies, taking into account national legislation.
For the situation when the device itself is not assessed for safety and/or performance, please refer to question 4.
Article 74(1) is only applicable to devices that are appropriately CE-marked at the time of the clinical investigation and the device must be used within the intended purpose as defined by the manufacturer. Note that modifications of the device may render the CE mark invalid.
12. How can a sponsor assess if the planned use of a device in the clinical investigation is covered by the intended purpose?
In order to assess if the use of a device in a clinical investigation is within its intended purpose, as defined by the manufacturer, first determine the device’s intended purpose, which could be identified by reviewing the instructions for use, and if available, the following documents:
- The EU declaration of conformity;
- The labelling (18) supplied by the manufacturer;
- Where applicable, the EU conformity certificate (19) for the device;
- The clinical evaluation plan;
- The clinical evaluation report;
- Where applicable, the Summary of Safety and Clinical Performance (SSCP).
The next step is to determine how the device will be used in the clinical investigation:
- Review the clinical investigation plan to determine the details of the planned use of the device. Details to review include the target population, the indications/contraindications, anatomical location where the device will used, the duration of use, the planned procedures, and the planned users.
- Check if the intended user in the clinical investigation will use the device as stated in the instructions for use.
From these documents, compare the intended purpose with how the device will be used in the clinical investigation and assess if these are aligned. An investigational device can only be considered to be used within its intended purpose if the planned use of the device during a clinical investigation is aligned with the device’s intended purpose.
13. What is considered burdensome or invasive?
Where the investigation would involve submitting subjects to procedures additional to those performed under the normal conditions of use of the device and those additional procedures are invasive or burdensome, the sponsor shall notify the Member States concerned at least 30 days prior to its commencement, in accordance with Article 74(1) of the MDR.
Additional procedures which are burdensome can include a wide variety of different interventions, this may include procedures which may cause pain, discomfort, fear, potential risks or complications/side-effects, disturbances of lives and personal activities, or otherwise unpleasant experiences. It is mostly determined from the perspective of the person bearing the burden.
Additional procedures which are invasive include (but are not limited to) penetration inside the body through the surface of the body, including through mucous membranes of body orifices, or penetration of a body cavity via a body orifice.
The understanding of what is considered to be invasive or burdensome is expected to develop over time. Sponsors are encouraged to document their assessment whether the additional procedures imposed by the clinical investigation plan are considered as burdensome and/or invasive, and where appropriate, contact the relevant authority in the Member State(s) to discuss cases where the sponsor is uncertain.
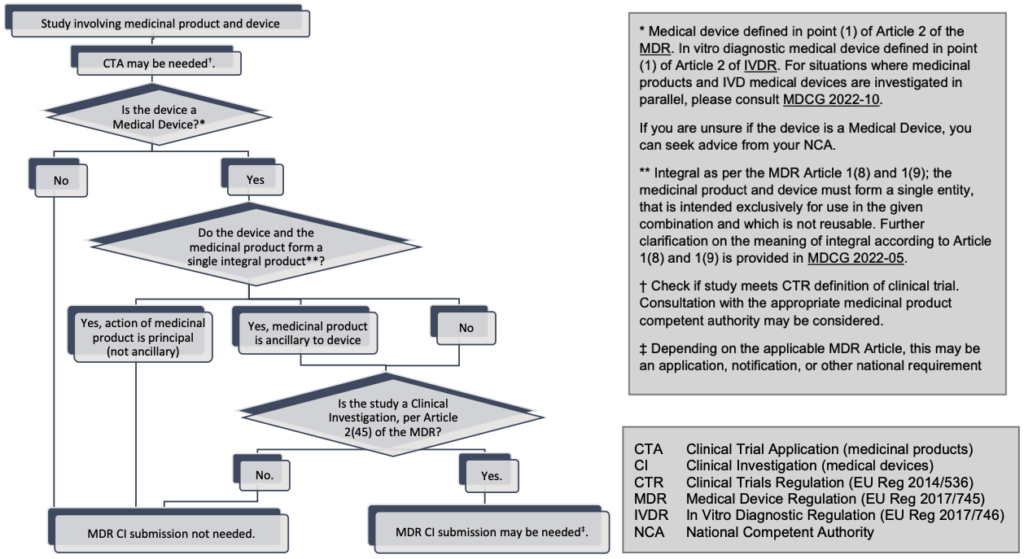
14. Are clinical studies of combinations of medical devices and medicinal products subject to the MDR requirements for clinical investigations?
Devices incorporating medicinal substances
Per Article 1(8) of the MDR, a device that, as an integral part, incorporates a substance which, if used separately, would be considered to be a medicinal product (20) and that has an action ancillary to that of the device shall be assessed and authorised in accordance with the MDR. Clinical studies of such devices that fulfil the definition of clinical investigation in Article 2(45) of the MDR are subject to the MDR requirements for clinical investigations.
However, if the action of the medicinal substance is principal and not ancillary to that of the device, the integral product shall be governed by Directive 2001/83/EC (21) or Regulation (EC) No 726/2004 (22) as applicable. In that case, the relevant general safety and performance requirements set out in Annex I of the MDR shall apply as far as the safety and performance of the device part are concerned. Clinical studies of such integral products may be considered clinical trials regulated by the Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR) (23).
Devices intended to administer medicinal products
Per Article 1(9) of the MDR, any device which is intended to administer a medicinal product (24) shall be governed by the MDR. Clinical studies of such devices that fulfil the definition of clinical investigation in Article 2(45) of the MDR are subject to the MDR requirements for clinical investigations.
However, if the device intended to administer the medicinal product and the medicinal product are combined in such a way that they form a single integral product which is intended exclusively for use in the given combination, and which is not reusable, that single integral product shall be governed by Directive 2001/83/EC (21) or Regulation (EC) No 726/2004 (22) as applicable. In that case, the relevant general safety and performance requirements set out in Annex I of the MDR shall apply as far as the safety and performance of the device part are concerned. Clinical studies of such integral products may be considered clinical trials regulated by the Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR) (23).
The flowchart in Annex III helps determine when the MDR requirements on clinical investigations are applicable for clinical studies of medical device and medicinal product combinations.
15. Which regulatory pathway shall a sponsor follow in order to conduct a clinical study (investigation) to collect clinical data both for a medicinal product and a device that will be used to administer the medicinal product, but it is not a single integral product?
The medicinal product and the medical device studied in a situation where they are not a single integral product at the time of administration, are governed by two different legal frameworks.
This applies even if they are co-developed and/or intended to be used in a specific combination.
Thus, a clinical study (6) which is collecting data for both a device and a medicinal product will have to comply with the regulatory requirements in relation to clinical investigations of medical devices (in the MDR) as well as the regulatory requirements for clinical trials of medicinal products, which are governed by the Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR).
If data on the device are being collected for conformity assessment purposes, Article 62 of the MDR applies.
16. Is there a common EU procedure for combined studies with devices and medicinal products?
No, clinical investigations of devices are regulated by the MDR, while clinical trials of medicinal products are regulated by the Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR). There is no dedicated procedure for combined studies foreseen in the regulations and therefore, currently, no harmonized procedure is in place. Therefore, the clinical study (6) has to be designed so that it fulfils both regulations. In administrative terms, combination studies have to be submitted as a notification or application for a device clinical investigation under the MDR and as a medicinal product clinical trial under the CTR. However, the study documentation required for submissions (if applicable) may be partially overlapping. Please note that each application must be sufficient in itself and cannot refer to documentation found in in other applications, as they are handled in separate systems. Sponsors of these clinical studies are encouraged to consult national guidance available via National Competent Authorities websites.
The clinical study (6) has to have the adequate design to fulfil the purpose of both legislations as intended. In administrative terms, the two regulatory pathways must be followed. Therefore, in relation to the device, Articles 62 or 82 of the MDR should be foreseen.
17. According to the MDR, are usability tests considered clinical investigations?
In the European standard EN 62366-1 (25):
- Usability is defined as the characteristic of the user interface that establishes effectiveness, efficiency, ease of user learning and user satisfaction in the intended use environment (section 3.16)
- Note 1 to entry: All aspects of usability, including effectiveness, efficiency and user satisfaction, can either increase or decrease safety of a medical device.
- Usability test is defined as a method for exploring or evaluating a user interface with intended users within a specified intended use environment (section 3.19)
- User interface is defined as the means by which the user and the medical device interact (section 3.26)
- Note 1 to entry: Accompanying documentation is considered part of the medical device and its user interface.
- Note 2 to entry: User interface includes all the elements of the medical device with which the user interacts including the physical aspects of the medical device as well as visual, auditory, tactile displays and is not limited to a software interface.
- Note 3 to entry: For the purposes of this standard, a system of medical devices can be treated as a single user interface.
- User is defined as person using, i.e. operating or handling, the medical device (section 3.24)
- Note 1 This includes, but is not limited to, cleaners, maintainers and installers.
- Note 2 Patients or other laypersons can be users.
- Use environment is actual conditions and setting in which users interact with the medical device (section 3.20)
- Note 1 to entry: The conditions of use or attributes of the use environment can include hygienic requirements, frequency of use, location, lighting, noise, temperature, mobility, and degree of internationalization.
Usability is created by characteristics of the user interface that facilitate use, i.e., to make it easier for the user to perceive information presented by the user interface, to understand and to make decisions based on that information, and to interact with the medical device to achieve specified goals in the intended use environments. Many of these factors can influence safety and performance (26) to various extents.
Manufacturers should document the usability of a medical device, and various types of usability test may be conducted for this purpose. Depending on the overall test design, usability testing may or may not fall under the definition of clinical investigation in Article 2(45) of the MDR (refer also to question 3).
Factors to be considered when determining whether a usability test is also a clinical investigation should include the scope and purpose of testing, as well as how the human subjects are exposed to the device. Note that prototypes or parts of a future device may have a medical purpose which is evaluated in the usability test and they would thus be considered as investigational devices, whereby the test could be a clinical investigation, in particular when the usability test involves exposing users to risk related to the device or where poor usability may lead to patient or user safety risks.
During device development it is recommended to design and conduct usability tests with limited human exposure to device related risks, in order to be able to adequately reduce the risks prior to the exposure to the device which is necessary for investigating device performance and/or safety in a subsequent clinical investigation.
Manufacturers should document (as part of the technical documentation) their justification of why a particular usability test falls outside the definition of a clinical investigation if human subjects are involved.
Typically, a usability test that fulfils the definition of clinical investigation falls under Article 62(1) of the MDR, but there may be situations where Article 82 of the MDR could be relevant.
18. May a retrospective clinical study fall under the definition of a clinical investigation?
A clinical investigation is defined in the MDR as “any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device”. In retrospective studies the involvement of human subjects and their exposure to the device precedes the study itself as data have already been generated. This implies that retrospective analysis of such data has no impact on patient management and the study does not introduce any additional risks for patients. The study may involve analysing ‘personal data’ which according to GDPR (27) means any information relating to an identified or identifiable natural person (‘data subject’) but the study per se does not mean involving (exposing) human subjects to a device.
If performance and/or safety of the device are analysed in the study retrospectively, separately from the decision to use the device, the study should not be considered as a clinical investigation according to the MDR, however there may be national provisions which need to be taken into consideration.
Data from such studies may form part of the clinical evaluation in appropriate circumstances.
19. When the objective of a clinical study is not specifically to evaluate a device, but the device is necessary for the purpose of the study (e.g. study of a physiological process, or a medical method or a surgical technique or rehabilitation technique) what regulatory aspects related to the device must the sponsor take into account?
Any device which is used for medical purposes as per Article 2(1) in research involving human subjects needs to comply with the requirements of the MDR that are applicable to it.
This means that the device must be:
- CE-marked according to MDD or AIMDD (have been acquired with a valid CE-marking), or
- CE-marked according to the MDR, or
- an “in house” device that fulfils the requirements of Article 5.5 in the MDR, or
- custom made device compliant with Article 52(8) of the MDR or
- an investigational device per Article 2(46) of the MDR used in a clinical investigation following the requirements outlined in either Article 62, 74 or 82 of the MDR. (28)
For devices lacking CE-marking, the only option available may be to design the study as a clinical investigation of the device and seek the necessary approvals. Although the main research purpose might not be to study the performance and/or safety of the device, these could be made secondary objectives. National legislation may provide further options.
Use of CE-marked devices outside their intended purpose in a clinical research setting should be carefully considered from a risk perspective, and may not always be appropriate, depending on the degree of deviation from the intended purpose stated by the manufacturer.
20. Who is responsible for determining the correct regulatory pathway for a clinical investigation?
It is the sponsor’s responsibility to determine the correct regulatory pathway for their clinical investigation. Guidance is provided in this document, but the MDR and national legislation contain the legally binding requirements.
Note that the MDR introduces a requirement for manufacturers to have access to a Person Responsible for Regulatory Compliance (Article 15 of the MDR).
Sponsors are encouraged to document their assessments and choices of regulatory pathways.
If the sponsor is uncertain about which route to apply for a particular clinical investigation, the National Competent Authority may be consulted.
21. What are the safety reporting requirements for clinical investigations?
For clinical investigations conducted according to article 62.1 and 74.1 the requirements for safety reporting will depend on whether you are using the investigated device within its intended purpose:
- If the investigated device is not CE marked, or is CE marked but will be used outside its intended purpose, the provisions on safety reporting laid down in Article 80 of the MDR shall apply.
- If the investigated device is CE marked and will be used within its intended purpose, the provisions on vigilance laid down in Article 80(5-6) and Articles 87 to 90 of the MDR and the acts adopted pursuant to Article 91 of the MDR shall apply for PMCF investigations.
Please refer to MDCG 2020-10/1 ‘Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745’ for further guidance (29).
Further, national CA websites should be consulted for national guidance on safety reporting, in particular for clinical investigations under article 82.
22. Does a manufacturer of devices without a medical purpose included in Annex XVI of the Regulation (EU) 2017/745 (MDR) have to conduct clinical investigations?
To ensure the same level of protection of consumers for some products which could be similar to medical devices but without a medical purpose, devices described in Annex XVI to the MDR have to comply with the applicable general safety and performance requirements. Clinical evaluations of those products shall be based on relevant data concerning safety, including data from post-market surveillance, PMCF, and, where applicable, specific clinical investigations. Clinical investigations shall be performed for those products unless reliance on existing clinical data from an analogous medical device is duly justified (30). Please refer to MDCG 2020-5 Clinical Evaluation – Equivalence, section 4 (f) for further guidance (31).
23. What procedure applies for clinical investigations of custom-made devices or in-house manufactured devices?
Custom-made devices are defined in Article 2(3) of the MDR.
In-house manufacturing, modifying and use of devices within health institutions is provided for in Article 5(5) of the MDR.
The relevant general safety and performance requirements set out in Annex I of the MDR apply to both of these device types. As such, clinical investigations may be undertaken with respect to these device types, and they may fall under Article 62 or 82. See Annex I of this document for further information.
24. Are coordinated assessment procedures in accordance with Article 78 of the Regulation (EU) 2017/745 (MDR) available?
For the time being, no coordinated procedure is available. When EUDAMED will be available and provide the functionality for applications under Article 78 of the MDR, some Member States will contribute to a voluntary test to start a co-ordinated assessment of multi-national studies.
Application content
25. What documents do I have to submit with an application for a clinical investigation that follows article 62(1) of the MDR?
The documentation to be provided is listed in chapter II in Annex XV of the MDR.
The guidance document MDCG 2021-08 on clinical investigation application/notification documents (32) provides templates for clinical investigation application/notification created to support clinical investigation procedures with respect to the MDR. In addition, it is necessary to visit the website of the relevant competent authority in the country where the clinical investigation is to be conducted, to check for national requirements regarding application content.
Additional, supporting documents may be provided in order to show compliance with applicable requirements and to provide sufficient information on the proposed study and investigational product(s) for the assessors to make a decision.
26. What documents do I have to submit for a clinical investigation that follows article 74(1) of the MDR?
Article 74(1) of the MDR points out that the same documentation has to be provided with a notification as for an application, see chapter II in Annex XV of the MDR.
The guidance document MDCG 2021-08 on clinical investigation application/notification documents (35) provides templates for clinical investigation application/notification created to support clinical investigation procedures with respect to the MDR. In addition, it is necessary to visit the website of the relevant competent authority in the country where the clinical investigation is to be conducted, to check national requirements regarding the notification content.
27. What documents do I have to submit for a clinical investigation that follows article 82 of the MDR?
Article 82 states minimum requirements for those clinical investigations that are not performed pursuant to any of the purposes listed in Article 62(1). These clinical investigations shall comply with the provisions of Article 62 (2) and (3), points (b), (c), (d), (f), (h), and (l) of Article 62(4) and Article 62(6).
It is not possible to provide general guidance on submission requirements for these studies, as the submission requirements for studies according to Article 82 of the MDR are subject to national provisions. Thus, it is necessary to check the website of the relevant competent authority and/or the relevant ethics committee in the country where the clinical investigation is to be conducted, to obtain information on national requirements.
28. What is the expected content of the Investigator’s Brochure (IB)?
The purpose of the IB is to provide the investigators with sufficient data on safety and performance of the investigational device, from pre-clinical testing or clinical investigations to justify human exposure to the investigational device.
Section 2 in chapter II of Annex XV of the MDR lists the required content of the IB. In addition, it is recommended that the annex B of ISO 14155:2020 (33) is adhered to.
For further guidance, consult MDCG document “Detailed guidance on content of the Investigator’s Brochure for clinical investigations of medical devices”.
29. What is the expected content of the Clinical Investigation Plan (CIP)?
The CIP (34) shall clearly outline the objectives and endpoints of the clinical investigation. The
proposed design shall be adequately justified based on scientific and ethical principles. Section 3 in chapter II of Annex XV of the MDR lists the required content of the CIP. In addition, it is recommended that the annex A of ISO 14155:2020 (35) is adhered to.
For further guidance, consult MDCG document “Detailed guidance on content of the Clinical Investigation Plan for clinical investigations of medical devices”.
Modifications to clinical investigations
30. How is a substantial modification defined?
A substantial modification of a clinical investigation is a change to the clinical investigation which is likely to have a substantial impact on the safety or health or rights of the subject, or on the robustness or reliability of clinical data generated by the investigation. Modifications of the clinical investigation plan (CIP), investigators brochure (IB), the subject information sheet and other clinical investigation documentation may or may not be considered as substantial modifications.
Sponsors should also take into consideration the fact that some modifications may seriously impact the design or scientific outcome of the clinical investigation and may require the initiation of a new clinical investigation.
The procedure to notify a substantial modification is further described in Article 75 of the MDR. A non-exhaustive list of modifications that may be interpreted as substantial can be found in Annex II of this document.
It is the sponsor’s responsibility to determine if the modification is substantial or not. However, if the sponsor is uncertain about the impact for a particular modification of a clinical investigation, the National Competent Authority may be consulted prior notification.
31. When can a sponsor submit a substantial modification notification?
A notification of substantial modification may be submitted in accordance with Article 75 of the MDR (36) as soon as a clinical investigation is allowed to commence in accordance with the MDR.
Moreover, it is not recommended to submit a substantial modification while assessment of an already submitted substantial modification is still ongoing. It is also important to consider whether national procedures may apply regarding modifications to clinical investigations. (37)
32. Is a change to the investigational device to be considered as a substantial modification to the clinical investigation or does it lead to the submission of a new clinical investigation application?
In general, a change to the investigational device is a substantial modification of the clinical investigation. As an example, a change to the device which alters the risk profile or adds new risks is likely to have a substantial impact on the safety or health or rights of the subject and, thus, is a substantial modification.
Some modifications to the investigational device are so extensive in nature that a new clinical investigation should be applied for. Modifications to the device which alter the suitability of the clinical investigation design to provide evidence for the safety, performance or clinical benefit of the device, may result in refusal of the modification and the submission of a new clinical investigation application may be required.
In order for the data resulting from a clinical investigation to be relevant for a certain device, in a situation where several versions of the device have been used during the clinical investigation, the aspect of equivalence may need to be considered.
MDCG 2020-5 provides guidance on equivalence in relation to clinical evaluation. This guidance may support sponsors in determining whether a modification of a device could be done within the same clinical investigation, or if a new clinical investigation application should be submitted. Member States will assess on a case-by-case basis if a change to the investigational device alters the suitability of the clinical investigation design to provide evidence for the safety, performance or clinical benefit of the device. If the modified investigational device is not equivalent to that one object of the initial application, according to MDCG 2020-5, a new clinical investigation may be required.
33. According to Article 75 of the MDR, if a sponsor intends to introduce modifications to a clinical investigation which are substantial, the sponsor has to notify the Member State within ‘one week’. From which point in time does this ‘one week’ start?
The ‘one week’ period starts from the date when the relevant documents (such as clinical investigation plan, investigator brochure, subject information sheet and informed consent form) are issued in an updated version.
It is acknowledged that changes to e.g. a CIP may require subsequent changes to other documents such as patient information, and that these changes may be done on a different date. Such changes can be collected and submitted together when the last affected document is issued but note that the implementation of the changes to the clinical investigation can not be done until the deadline in Article 75 of the MDR has expired or an authorisation letter is issued by the Competent Authority and/or Ethics Committee if this is required according to national provisions.
34. Can the sponsor start to implement the substantial modification after 38 days of the notification date to the Member State?
Yes, if the sponsor has not heard from the Member State after 38 days the substantial modification may be implemented, provided that an Ethics Committee in that Member State has not issued a negative opinion in relation to the substantial modification. This 38-day period, which starts when all the relevant documentation with clearly identifiable changes have been submitted in their entirety, may be extended by a further 7 days in order to consult with experts. Member States will notify the sponsor if such a consultation is taking place. The substantial modification can be implemented sooner if the Member State has authorised the substantial modification.
If the Member State has sent a request for information, there may be, depending on national provisions, a clock-stop, as long as the Member State has not received the additional information.
Further, the sponsor needs to ensure that updated documents in relation to a substantial modification are submitted to both the Competent Authority and the Ethics committee where required. Note that there may be national provisions in place regarding e.g. the notification and review of substantial modifications to Ethics committees.
35. What notification requirements apply to non-substantial modifications?
Article 75 of the MDR does not describe how sponsors or authorities shall deal with non-substantial modifications. Once EUDAMED is available, sponsors are expected to keep the information in the database up to date in accordance with Article 70(2) of the MDR. However, in the absence of EUDAMED Member States have not yet harmonised their approach, and it is thus necessary to check the national requirements.
Note that there may be national provisions in place regarding e.g. the notification and review of non-substantial modifications to Ethics committees.
Timeline considerations for clinical investigations
36. Which date is considered as the start of the clinical investigation?
Reporting of the clinical investigation start date is not explicitly required by the MDR, but in some Member States it is required by national legislation to report this to the relevant Authority.
Further, the start date of a clinical investigation should be indicated in EUDAMED (once available) to disseminate relevant information to the public and for Competent Authority inspection planning purposes. The start date of a clinical investigation should be described in the clinical investigation plan. In general it is considered to be, the first act of recruitment in the clinical investigation in a Member State. The first act of recruitment should be specified by the sponsor and could be, for example, the date of initiation of the clinical investigation in the first site or the date when the first investigation-specific advertisement is published. In any case the clinical investigation cannot start earlier than the authorisation date (or commencement date notified for PMCF clinical investigations) or not later than the date recruitment starts.
37. Which date is considered as the end date of a clinical investigation?
As stated in Article 77(2) of the MDR, the end of a clinical investigation shall be deemed to coincide with the last visit of the last subject unless another point in time for such end is set out in the clinical investigation plan (for example site closure occurring after the last visit of the last subject).
38. Does the sponsor have to notify the end of the clinical investigation when it is concluded in one or more Member States, or when the overall clinical investigation is completed globally?
According to Article 77(3) of the MDR the sponsor shall notify each Member State in which a clinical investigation was being conducted of the end of that clinical investigation in that Member State. That notification shall be made within 15 days of the end of the clinical investigation in relation to that Member State.
If a clinical investigation is conducted in more than one Member State the sponsor shall also notify all Member States in which that clinical investigation was conducted when the clinical investigation is completed in all Member States. That notification shall be made within 15 days of the end of the clinical investigation in the last Member State.
If the clinical investigation is still ongoing in one or more third countries when the end of the clinical investigation in the EU is reported, this will impact the sponsor’s ability to provide a clinical investigation report of the overall study (i.e. fulfil the reporting requirements of Article 77(5) of the MDR). Therefore, the sponsor should inform the concerned Member States of the expected end of study globally if this does not coincide with the end of study in the EU. Sponsors are encouraged to notify the Member States concerned to confirm the actual end of study globally, once reached.
39. When does the sponsor have to notify an early termination of a clinical investigation?
In case a clinical investigation is temporarily halted or terminated early, the sponsor has to inform the Member State concerned within 15 days, or within 24 hours if based on safety grounds. This reporting timeline should be counted from the sponsor’s decision to halt/terminate the clinical investigation.
In case of restart after temporarily halted study, a notification will be made to the CA. In case of temporarily halt on safety grounds, this notification should be submitted as a substantial modification.
Note that if inclusion of new subjects is halted in a country before the expected number of subjects in that Member State was reached, and this is due to the achieved recruitment of the total number of subjects globally, this is not considered as early termination.
40. When does the sponsor have to submit the clinical investigation report with the results?
Irrespective of the outcome of the clinical investigation, within one year of the global end of the clinical investigation the sponsor shall submit to all the Member States in which a clinical investigation was conducted a clinical investigation report as referred to in Section 2.8 of Chapter I and Section 7 of Chapter III of Annex XV to the MDR.
Following a decision of an early termination or temporary halt, a clinical investigation report is required to be submitted within 3 months to all Member States in which the clinical investigation was conducted. Sponsors are expected to submit a risk analysis concerning any safety grounds related to the temporary halt.
In the event that the clinical investigation is restarted within three months of the temporary halt, the sponsor does not have to submit a clinical investigation report until the clinical investigation has been completed. The final clinical investigation report should include detail with respect to the temporary halt.
41. For how long must study documentation be retained?
It is stated in section 3 chapter III (covering sponsor’s obligations) in Annex XV of the MDR that the documentation mentioned in annex XV shall be kept for a period of at least 10 years after the clinical investigation with the device in question has ended, or, in the event that the device is subsequently placed on the market, at least 10 years after the last device has been placed on the market. In the case of implantable devices, the period shall be at least 15 years.
Note that the phrase ‘placed on the market’ refers to each single individual device, i.e. the reference is to the last individual device transferred from the manufacturer to another party. (38) This means that in practice documentation has to be stored for an extended period of time.
Clinical investigation reports
42. What shall be the content of the clinical investigation report?
The minimum requirements for content of the clinical investigation report (which will be made public according to Article 77 of the MDR) are defined in Chapter III point 7 of Annex XV to the MDR. The standard ISO 14155:2020, Annex D also has information which is relevant regarding the content of a clinical investigation report.
It is important to note that the summary of serious adverse events, adverse device effects and device deficiencies should only present aggregated information related to these events.
Descriptions of single events or line listings with direct or indirect personal data may jeopardise subject privacy and should be avoided in a report which will be made public.
To put the information that is required (minimum content) in a relevant context and enhance the understanding of the clinical investigation report, sponsors are encouraged to also include the following information in the report:
Clinical investigation background
Presentation of the context and reasons for conducting the clinical investigation.
Outcome measures
Description of the selected outcome measures and their relevance for the assessment of safety and performance of the investigational device.
Clinical investigation conduct
- Include information on the dates defining the periods of recruitment and follow-up of subjects in order to describe the time period during which the clinical investigation was conducted.
- Interventions: Precise details of the interventions intended for each group and how and when they were actually administered should be included in the report. State the precise dose (if relevant), treatment duration, control interventions and additional treatment for each of the groups.
Clinical investigation subjects
Baseline data (baseline demographic and clinical characteristics of each group) should be included. Also describe the flow of subjects through each stage (diagram, if appropriate).
For each group the numbers of subjects randomly assigned, receiving intended treatment, completing the clinical investigation, and analysed for the primary outcome should be stated.
Indicate the number of subjects in each group which have been included in each analysis and whether the analysis was by „intention-to-treat“ or “per protocol”.
Deviations and amendments
Deviations from the initial clinical investigation plan and a description of any CIP amendments should be described and justified.
43. Is there a template for the mandatory summary of the clinical investigation report referred to in article 77(5) of the MDR?
Yes, refer to the Commission Guidance on the content and structure of the summary of the clinical investigation report (2023/C 163/06) (39).
Arrangements for the transitional period
44. When will EUDAMED allow applications for clinical investigation?
The use of EUDAMED for Clinical Investigation and Performance Studies becomes mandatory at the end of the 6 months transitional period after the publication of a notice in the Official Journal of the European Union that EUDAMED has achieved full functionality following the outcome of an independent audit. It will not be possible to submit applications via EUDAMED before it is mandatory to use.
Link to the EUDAMED: https://ec.europa.eu/tools/eudamed
45. Until the clinical investigation module in EUDAMED is ready, how can the sponsor comply with the MDR?
Clinical investigation sponsors will not have the opportunity to register as actors in EUDAMED as of the date of application of the MDR, nor to submit applications or notifications via EUDAMED.
To submit an application:
All requested information to apply for or notify a clinical investigation should be submitted to the national competent authorities unless otherwise specified in the MS concerned. Check with the relevant National Competent Authority which system will be used for submission.
The Commission has a listing of the contact details for National Competent Authorities, which is available at: https://ec.europa.eu/health/sites/health/files/md_sector/docs/md_clinical_investigation_contact_points.pdf
To fulfil the safety reporting requirements of the MDR Article 80:
Please see MDCG 2020-10/1 ‘Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745’ for guidance.
46. What will happen to those clinical investigations that started prior to the date of application of Regulation (EU) 2017/745?
Clinical investigation that are currently being conducted with respect to Directive 93/42/EC and Directive 90/385/EC by the date of application of the MDR, can continue to be conducted. Nevertheless, serious adverse events (SAEs) and device deficiencies occurring after the date of application of the MDR, shall be notified to the MS according to the rules defined in Article 80 of the MDR.
To facilitate the transition and give time for sponsors to update Clinical Investigation Plans and procedures in clinical investigations a sponsor may continue to report all SAEs to National Competent Authorities until Eudamed reporting is mandatory. This applies only to studies which have started to be conducted in accordance with Article 10 of Directive 90/385/EEC or Article 15 of Directive 93/42/EEC prior to 26 May 2021. Please refer to MDCG 2020-10/1 ‘Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745’ for further guidance.
47. How should Article 120(11) of the MDR be interpreted – when is a clinical investigation to be considered started to be conducted in accordance with Article 10 of Directive 90/385/EEC or Article 15 of Directive 93/42/EEC?
As national interpretations and implementation of the directives in national legislation may differ between Member States, please check with the national competent authority in the Member State where the clinical investigation is to be conducted as to what they consider as started/starting date.
Legal representative
48. What is the role and responsibility of the sponsor’s legal representative?
In article 62(2) of the MDR it is stated that: Where the sponsor of a clinical investigation is not established in the Union, that sponsor shall ensure that a natural or legal person is established in the Union as its legal representative (40). Such legal representative shall be responsible for ensuring compliance with the sponsor’s obligations pursuant to this regulation, and shall be the addressee for all communications with the sponsor provided for in this Regulation. Any communication with that legal representative shall be deemed to be a communication with the sponsor.
Per the Regulation, the legal representative is responsible for ensuring compliance with the sponsor’s obligations pursuant to the MDR. It is not foreseen that this responsibility can be delegated back to the sponsor, nor to a CRO.
It is recommended that the sponsor issues a study specific power of attorney to the designated legal representative.
In order to enable the legal representative to ensure compliance with the sponsor’s obligations under the MDR it is recommended that a contract between the parties obliges
– the sponsor to provide the legal representative with all required information and
– the legal representative to immediately notify the sponsor in case (s)he becomes aware of any noncompliance with the Regulation.
If tasks are delegated by a legal representative to another legal or natural person, the responsibility to ensure compliance with the MDR still resides with the legal representative.
Any delegation of tasks related to sponsor’s obligations must be done in compliance with chapter III of annex XV to the MDR. If the contract(s) between the sponsor and other entities are related to the compliance of the sponsor’s duties according to the MDR, it is also the responsibility of the legal representative to ensure that these duties are well covered by the other entities.
It is acknowledged that responsibilities and tasks contracted to the legal representative may expand beyond the legal requirements of the MDR, which is a business decision between the parties. In situations where the legal representative also functions as a CRO, it is recommended that the agreement between the sponsor and the CRO clearly defines what responsibilities are assigned in terms of the legal representative capacity and what tasks are assigned to the CRO capacity.
49. How should a legal representative check the sponsor’s compliance with the MDR?
The legal representative is responsible for ensuring compliance with the sponsor’s obligations pursuant to the MDR. The MDR does not specify how a legal representative fulfils the legal obligations.
The legal representative may perform audits of the sponsor and its subcontractor(s), or check compliance by other means, according to prior agreement.
The legal representative would need permanent and integral availability and access to the technical documentation of the investigated device and clinical investigation documentation, in order to be able to ensure that this information has been drawn up and complies with the MDR.
For the legal representative it would be necessary to be aware of specific aspects of the clinical investigation, i.e. not only to check the availability of procedures on a general level., in order to be able to ensure that the clinical investigation is compliant with the MDR in practice.
It is necessary that the legal representative verifies, in parallel and/or in addition to the sponsor, all relevant documentation and keeps records of the verification activities performed. This verification is included in the legal representative’s responsibility for ensuring compliance with the sponsor’s obligations pursuant to the MDR.
However, this does not mean that the legal representative replaces the sponsor with regard to its obligations expressed in Chapter III, Annex XV, nor does it mean a participation in the evaluation/review of the data of each document.
50. Would it be sufficient that the legal representative is able to provide documents from the sponsor upon request by authorities?
It is expected that the legal representative has permanent and unlimited access to the complete and up-to-date investigation master file including, but not limited to, contracts and financial arrangements related to the clinical investigation, evidence that the investigational sites and other participating entities and persons are appropriately qualified, relevant communication which can support the demonstration of compliance of sponsor’s duties according to the MDR, monitoring reports and SAE reports. It should be specified in the contractual agreement between the sponsor and the legal representative how this is achieved.
Note that it is clearly stated in section 3, chapter III, Annex XV of the MDR that the documentation should be kept at the disposal of the competent authorities, even if the sponsor goes bankrupt or ceases its activity prior to the end of the specified retention period.
Further, sponsors and legal representatives should also consider the fact that securing storage on EEA and Türkiye territory may be critical in the event that something happens to the sponsor’s documentation.
Annex I: Clinical investigation under the MDR – regulatory pathway
Annex II: Non-exhaustive list of modifications that may be interpreted as substantial
Amendments related to the clinical investigation plan or subject information
1. Change to a primary or secondary endpoint;
2. Use of a new mode of measurement for the primary endpoint;
3. A change of clinical investigation design which is likely to have a significant impact on the statistical analysis or the benefit/risk assessment;
4. A change in the definition of the end of the clinical investigation;
5. A modification of the duration of treatment and/or the follow up of subjects;
6. Changes in the number of scheduled subject visits;
7. Change of a diagnostic or other assessment procedure which is likely to have a
significant impact on the safety of the subject or the scientific value of the clinical data collected in the clinical investigation;
8. Changes to the data monitoring committee which may affect, for example, the safety evaluation, or the independence and impartiality of the committee;
9. Amending the number of subjects to be included in the clinical investigation, either due to an adaptation of the sample size calculation or to maintain a previously defined sample size calculation due to an increased unanticipated dropout rate;
10. Addition of an interim analysis not planned in the initial CIP;
11. Deletion of an interim analysis;
12. Change of safety criteria to modify or interrupt treatment;
13. Content change in the subject information sheet and informed consent forms, or other information provided to the subject;
14. Change of inclusion or exclusion criteria if these changes are likely to have a significant impact on the safety of the subject or scientific value of the clinical data collected in the clinical investigation.
Amendments related to the benefit/risk of the clinical investigation
15. New preclinical or clinical data which is likely to impact on the benefit/risk assessment;
16. The revocation or suspension of the conformity assessment certificates related to the device under investigation.
Amendments related to the use of the investigational device
17. Change of treatment modalities (modification of procedure, techniques, instructions for use) of the investigated device;
18. The type and/or duration of the investigator’s training.
Amendments related to other information
19. A change of sponsor or the sponsor’s legal representative;
20. Change/addition of a clinical investigation site;
21. Change of manufacturer;
22. New insurance policy;
23. Change in compensation paid to subjects and/or investigators/site;
24. Change of /addition of new investigator(s).
Amendments related to the manufacturing process
25. Modification of the process of manufacturing, sterilization or packaging
Annex III: Does my combination product study require an MDR clinical investigation submission?
Footnotes
(1): Article 1(4) of the MDR.
(2): Note that in the EU regulation 536/2014 the term Clinical Study has a much narrower definition and is limited to studies involving medicinal products.
(3): This is also in contrast with the Note 1 to entry 3.8 in ISO 14155:2020 which states that ‘For the purpose of this document, ”clinical trial” or “clinical study” are synonymous with “clinical investigation”.
(4): Defined in Article 2(49) of the MDR as any individual, company, institution or organisation which takes responsibility for the initiation, for the management and setting up of the financing of the clinical investigation.
(5): Article 82(2) of the MDR, Article 70(7)(a) of the MDR.
(6): Please refer to the Introduction of this document for clarification on how the term ꞌclinical studyꞌ is used in this guideline.
(7): such as the General Data Protection Regulation (GDPR) Regulation (EU) 2016/679 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data.
(8): Article 2(45) of the MDR.
(9): Note that if an endpoint or objective in such a study is related to the assessment of the safety or performance of the device, the study would meet the definition of clinical investigation in Article 2(45) of the MDR.
(10): As specified in Article 5 of the MDR.
(11): Refer to question 2 regarding other applicable legislations.
(12): Defined in Article 2(22) of the MDR.
(13): For some medical devices, performance may relate to the user of the device.
(14): Defined in Article 2(52) of the MDR.
(15): Defined in Article 2(53) of the MDR.
(16): For certain investigations, in particular early studies of new/high risk devices it might be necessary from a safety point of view to limit the rate of enrolment (e.g.; the study design could include an evaluation of the first patient before next patient is treated, a phased approach, use of a run-in component).
(17): Clinical investigation of medical devices for human subjects – Good clinical practice (ISO 14155:2020)
(18): Article 2(12) of the MDR defines “intended purpose” as “the use for which a device is intended according to the data supplied by the manufacturer on the label, in the instructions for use or in promotional or sales materials or statements and as specified by the manufacturer in the clinical evaluation”.
(19): EU technical documentation assessment certificates, EU type-examination certificates or EU product verification certificates as appropriate.
(20): As defined in point 2 of Article 1 of Directive 2001/83/EC, including a medicinal product derived from human blood or human plasma as defined in point 10 of that directive
(21): Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use
(22): Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency
(23): Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC
(24): As defined in point 2 of Article 1 of Directive 2001/83/EC
(25): EN 62366-1 Medical devices – Part 1: Application of usability engineering to medical devices (IEC 62366-1:2015)
(26): The words ”and performance” have been added vs the text in standard EN 62366-1
(27): Article 4(1) Regulation (EU) 2016/679 of the European Parliament and of the Council of 27 April 2016 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data, and repealing Directive 95/46/EC (General Data Protection Regulation)
(28): Note that Member States may impose additional national requirements for clinical investigations falling under Article 82.
(30): Article 61(9) of the MDR
(33): Clinical investigation of medical devices for human subjects – Good clinical practice (ISO 14155:2020)
(34): May also be called “protocol”
(35): Clinical investigation of medical devices for human subjects – Good clinical practice (ISO 14155:2020)
(36): Please refer to Q30 of this document for further details.
(37): For example, national procedures relating to Ethics Committee opinions.
(38): This is eluded from the Blue Guide
(39): 2023/C 163/06
(40): Article 62(2) of the MDR allows Member States to choose not to apply the requirement for sponsors to have a legal representative in clinical investigations to be conducted solely on their territory, or on their territory and the territory of a third country, provided that they ensure that the sponsor establishes at least a contact person on their territory in respect of that clinical investigation who shall be the addressee for all communications with the sponsor.
Revision History
Redline Version