
MDCG 2020-3 Rev.1
Guidance on significant changes regarding the transitional provision under Article 120 of the MDR with regard to devices covered by certificates according to MDD or AIMDD
Disclaimer: This document is an interactive version of the original MDCG document. We will keep it up-to-date.
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.
MDCG 2020-3 revision 1 changes
MDCG 2020-3 revision 1 changes |
|---|
Adjustments all over the document to align it to Regulation (EU) 2023/607 and guidance MDCG 2022-2 |
1 Introduction
Article 120(3) of the Medical Device Regulation (EU) 2017/745 (MDR), last amended by Regulation (EU) 2023/607, states that devices which continue to comply with the AIMDD or MDD may be placed on the market or put into service until 31 December 2027 (1) or 31 December 2028 (2), as applicable, provided the conditions set out in Article 120(3c) MDR are fulfilled.
These devices are called ‘legacy devices’ and in line with MDCG Guidance Document 2021-25 (3), ‘legacy devices’ should be understood as devices, which, in accordance with the MDR’s transitional provisions, are placed on the market after the MDR’s date of application (i.e. 26 May 2021) if certain conditions are fulfilled. Those devices can be:
devices which are class I devices under Directive 93/42/EEC (MDD), for which an EC declaration of conformity was drawn up prior to 26 May 2021 and for which the conformity assessment procedure under the MDR requires the involvement of a notified body;
devices covered by a valid EC certificate issued in accordance with Directive 90/385/EEC (AIMDD) or the MDD prior to 26 May 2021. (4)
The conditions are set out in Article 120(3c) MDR and include, among others, that legacy devices must continue to comply with the AIMDD/MDD (5), as applicable, and that there are no significant changes in the design or intended purpose of the device (6). Therefore, it is important for manufacturers and notified bodies to have a clear understanding as to what changes to design or intended purpose would be considered ‘significant’.
It is essential for legacy devices that their certificates remain valid (7) following changes that are not significant with regard to design or intended purpose and that the required appropriate surveillance is carried out (8).
The first revision of this guidance document MDCG 2020-3 on significant changes regarding the transitional provisions under Article 120(3) MDR takes into account the experience gained with the application of the original version so far, is aligned with the equivalent guidance for IVDs, MDCG 2022-6 of May 2022 (9) and adjusted to Regulation (EU) 2023/607.
2 Scope
This guidance document is intended to provide clarification on the concept of ‘significant changes in the design and intended purpose’ under Article 120(3c), point (b) MDR. It concerns manufacturers of legacy devices.
This guidance document does not elaborate on the process for manufacturers’ submission and notified bodies’ assessment of changes to the approved design (10) or substantial changes to the approved quality system or the product-range covered. (11)
3 Changes to Directive certificates
It is important to highlight that no issuing of new AIMDD/MDD certificates is allowed under Article 120(3c), point (b) MDR (12) (see also section 4.1). In particular, if the manufacturer wishes to make a ‘significant change in design or intended purpose’ within the meaning of Article 120(3c), point (b) MDR, the implementation of such a change would prevent the manufacturer from placing the device on the market under the AIMDD/MDD in accordance with that provision. (13)
4 Assessment whether changes are ‘significant changes in the design or intended purpose’ in accordance with Article 120(3c), point (b) MDR
4.1 General
In order to benefit from the transition periods provided by Article 120(3) MDR, legacy devices may not undergo any significant change in the design or intended purpose after the date of application of the MDR, i.e. 26 May 2021.
A significant change in the design or intended purpose consist of two cumulative elements:
there is a change in the design or intended purpose, and
the change is significant.
That means that changes that do not concern the design or intended purpose are out of scope of Article 120(3) MDR. Equally, changes that concern the design or intended purpose only fall under Article 120(3) MDR if they are considered ‘significant’.
Changes in order to comply with other Union legislation are also out of scope even if they relate to changes in the design, only if the risk/benefit ratio of the device is not negatively affected.
The manufacturer is responsible for providing evidence and justification that a change does not affect the design or intended purpose, or, in case the change affects design or intended purpose of the device, that it is non-significant, which should be assessed case-by-case. The outcome of the assessment should be documented and made available to a competent authority when requested.
If a change is not a significant change in design or intended purpose, the implementation of such a change is allowed during the transitional period without the need for certification under the MDR. In such cases, the manufacturer is required to comply with the documentation requirements of the AIMDD/MDD, i.e. the updated technical documentation must allow assessment of the conformity of the product with the applicable requirements.
Additional considerations for devices covered by a certificate issued by a notified body
In line with the agreed arrangements for notification of changes between the manufacturer and the notified body according to the AIMDD/MDD (e.g. contractual relationships, approved procedures (14)), changes and their implementation are to be verified by the notified body as part of the surveillance activities, or following a manufacturer’s submission for prior approval. The outcome of this verification will determine whether a certificate in accordance with the AIMDD/MDD remains valid according to Article 120 MDR. In case of doubt as to whether a given change, which concerns the intended purpose or the design is significant, manufacturers should ask their notified body.
The certificate should not be amended. However, the notified body that issued the certificate in accordance with the AIMDD/MDD may confirm in writing, after having reviewed the manufacturer’s description of the (proposed) change, that the implementation of the change does not represent a significant change in design or intended purpose under Article 120(3c), point (b) MDR and that the related AIMDD/MDD certificate remains valid until the end of the transition period (15). Such written confirmation corrects or complements information on an existing certificate but does not represent the issuance of a “supplemented certificate”, since this is prohibited as mentioned in Section 3. In case of requests from authorities, the manufacturer should number such letters (16) received from the notified body and submit them together with the certificate.
Where the conformity assessment procedure under the AIMDD/MDD for a given legacy device requires the quality management system (QMS) of the manufacturer to be approved by a notified body, the manufacturer must observe that the conditions for which the certificate was granted are maintained. In any case, changes to the QMS continue to be subject to the agreed notification procedure between manufacturer and notified body.
4.2 Changes not concerning the design or intended purpose
Changes concerning the manufacturer’s organisation (administrative changes) or changes concerning the manufacturing process should generally not be considered changes in the design or intended purpose within the meaning of Article 120(3c), point (b) MDR, even if they need to be reflected in the information to be supplied with the device (e.g. label or instructions for use).
This includes for example:
- changes of the manufacturer’s name, address or legal form, including a merger or acquisition involving the manufacturer (17);
- changes in relation to the authorised representative;
- relocation or addition of new manufacturing sites, including when it affects subcontractors or suppliers;
- changing the supplier of a material, substance (18) or component, provided the specifications of the new material, substance or component do not change;
- adding or replacing a new material number for logistic reasons without changing the material;
- new process validation as part of manufacturing improvements or scale-up of manufacturing.
Changes of the QMS, such as changes in the monitoring and control of production and operations environment, generally do not impact the design or intended purpose either, provided that the conditions for which the conformity assessment certification was granted are maintained. In any case, changes continue to be subject to the agreed notification procedure between manufacturer and notified body.
4.3 Changes in the design or intended purpose
4.3.1 Design and intended purpose
As legacy devices need to be in compliance with AIMDD/MDD, the benchmark for determining their design and intended purpose, as well as any possible change, should be the AIMDD/MDD respectively.
While the AIMDD/MDD do not define the “design” of a device, the “intended purpose” means “the use for which the device is intended according to the data supplied by the manufacturer on the labelling, in the instructions for use and/or in promotional materials” (Article 1(2) letter (f) AIMDD and Article 1(2) letter (g) MDD).
4.3.2 Significance of changes
To facilitate a harmonised judgement of the significance of a change, this guidance document provides several flowcharts in the Annex. The assessment of a proposed change using the main flowchart and any of the applicable sub-charts is intended to assist manufacturers, notified bodies and market surveillance authorities in deciding whether or not a change of the design or intended purpose of the device is to be considered significant under Article 120(3c), point (b) MDR.
A change is considered a non-significant change of design or intended purpose per Article 120(3c) point (b) MDR if the answer to every question in a sub-chart leads to “non-significant change” also when returning to the main chart. On the contrary, if any sub-chart delivers the result “significant change”, the change being assessed is a “significant change in design or intended purpose” according to the Article 120(3c), point (b) MDR.
4.3.2.1 General considerations
As a rule, the following changes in design and/or intended purpose should be regarded as “non- significant”:
changes related to corrective actions (19) assessed and accepted by the competent authority of the Member State in which the manufacturer or its authorised representative has its registered place of business (see also Q&A 17 of the CAMD’s FAQ – MDR Transitional provisions (20)) regarding design changes – the same approach should apply also to changes related to the intended purpose;
correction of spelling mistakes or merely editorial changes of the information to be supplied with the device (e.g. label or instructions for use);
updates of the information to be supplied with the device (e.g. label, instructions for use or implant card) if they are required by EU legislation other than the MDR (e.g. CLP Regulation (EC) No 1272/2008 on classification, labelling and packaging of substances and mixtures (21) read in conjunction with Annex I, section 7.5 MDD), are mere clarifications and do not adversely affect the devices’ safety and performance in relation to existing or new risks;
clarifications of intended purpose, population, clinical application in the information to be supplied with the device in line with the original certification.
4.3.2.2 Changes in the intended purpose – Chart A
Regarding changes of the intended purpose the following principles should apply (see chart
A):
Non-significant change:
-Limitation of the intended purpose (see Q&A 17.3 of the CAMD’s FAQ – MDR Transitional provisions20), such as:
restriction or deletion of certain indications;
restriction or deletion of certain applications, e.g. anatomical site, delivery pathway or deployment method;
restricting the target population.
Significant change:
-Extension of the intended purpose, such as:
- additional or new indications;
- additional or new clinical conditions.
-New user or patient population, such as:
- additional or new target population;
- additional or new user (e.g. change from professional use to layman use).
-New way of clinical application, such as:
- additional or new applications (different stage or severity of disease);
- additional or new anatomical site;
- new delivery pathway or deployment method.
When assessing whether the intended purpose is being changed, changes in the label or instructions for use, as well as promotional materials, that are linked to the use for which a device is intended (e.g. limitations, warnings) should be considered.
4.3.2.3 Changes in the design – Charts B to E
Changes concerning software, substances or materials, or sterilisation also concern the design of the device. Specific flowcharts (C, D, E) are intended to assist in assessing whether changes in those areas should be considered significant changes in the design.
Regarding changes of the design the following principles should apply (see chart B):
Note: Change of a software shall be primarily evaluated per chart C. However, there may be cases where a major change of a software may be considered a change in built-in control mechanism or device’s operating principles and therefore are evaluated according to chart B.
Non-significant change:
-change of the design that does not alter:
- the built-in control mechanism,
- the device’s operating principle,
- the change of the source of energy, or
- the alarms systems and
which does not adversely affect the safety, performance or usability and negatively affect the risk/benefit ratio of the device.
Examples:
Change in Specification/Labelling:
change within the currently certified range (more narrow or detailed information), new article inside certified worst case or accepted bracket validations such as:
- new screw variant within current range of lengths and diameter;
- new catheter variant, with length and diameter within current range and worse case in sterilisation performance;
- new stent lengths which are intermediate between the previously certified stent lengths.
-change of colour of an assembly or device;
-change to the grip of a steerable ablation catheter to provide improved ergonomic comfort for the healthcare professional or aesthetic presentation of the device without changing the functionality;
-change to device housing that are minor reinforcement or attachments (handles/extra handles) or changes to supporting parts (for reduction of vibration);
-change to design for an easier operation, such as change to the thumb rest of injection syringe, change to lids or change to the mechanism of loading substances;
-change/modification of a connector to reduce misconnection and thereby improve safety of use;
-modification of a part (e.g. a connector) to adhere to a new or revised standard;
-removal of accessories (e.g. torque device) from a system (which is certified as a device) that are otherwise clinically readily available;
-formatting an existing IFU content into a newly created IFU, including reorganization of information;
-editorial labelling improvements to warnings and precautions to better communicate and clarify critical information to the end user;
-adding to an existing IFU a new list of accessories including new versions of an accessory assigned to the device;
-change of label or packaging material of a non-sterile device e.g. a change in packag- ing from one variant of polyethylene to another due to supplier rationalization or cost saving measures;
-change to primary package size within previously validated range and previously vali- dated sterilisation parameters;
-changes to outer packaging (e.g. size, material, layout) that do not adversely affect the stability, sterility or microbiological state of the device;
-extension of temperature or humidity range limitations for operation or storage;
-changes in reprocessing instructions, including increase of the number of allowed reprocessing cycles of a re-usable device in the related labelling, if achieved without changing the device design but provision of appropriate testing.
Note: These examples are valid only provided that the risk/benefit ratio of the device is not negatively affected.
Change of a component of a device (in particular electronics):
-change of a semiconductor/electronic component/electronic assembly, e.g. due to obsolescence;
-replacement of a semiconductor/electronic component/electronic assembly with the same specifications.
-replacement of a semiconductor/electronic component/electronic assembly with the same function, and requiring changes such as:
- re-layout of the printed circuit board (e.g. if not drop-in compatible);
- installation of a new firmware of software driver to operate the new semi-conductor;
- adjustments of the software operating the device to accommodate the new semiconductor to operate as intended;
- re-arrangement of interfaces.
-addition of a coating on a non-contacting patient printed circuit board assembly (PCBA) to improve electrical current insulation;
-replacement of alarm signal generation components, e.g. LEDs, speakers;
-change of size or geometrical shape of operation/alarm buttons;
-change of the audible alarm mode by an additional red light.
Note: These examples are valid only provided that the risk/benefit ratio of the device is not negatively affected.
Change to the source of energy:
-change of a battery type, e.g. from AA to 6LR61;
-change of battery chemistry, e.g. from Alkaline to Lithium Manganese Dioxide or from lead acid batteries to lithium iron phosphate (LiFePo) batteries;
-change from a battery to a rechargeable accumulator, if recharging takes place outside of the device;
-change of battery charger or supply cords (under same specification).
Note: These examples are valid only provided that the risk/benefit ratio of the device is not negatively affected.
Significant change:
-change of the design that alter:
the built-in control mechanism,
the device’s operating principle,
the change of the source of energy, or
the alarms systems.
Examples:
change from analogue to digital control;
change from manual to software driven device;
change to measuring function, wavelength or light emission;
replacement of PCB with new feature or change of control circuits;
change from energy source battery or accumulator to net-power operation of the device or vice versa; or from LED (Light-Emitting Diode ) to U.V rays;
reduction of battery operating time;
change of an advisor for the Insulin-bolus-volume in an Insulin pump;
silencing/removing/adding of an alarm system or handling of alarm situation.
-changes that may adversely affect the safety or performance and negatively affect the risk/benefit ratio of the device, even if they do not alter the built-in control mechanism, the device’s operating principle, the change of the source of energy or the alarm systems.
Examples:
-change to device dimensions or design characteristics outside of current specifications, such as:
- new stent lengths which are outside of the range of the previously certified stent lengths;
- addition of two or more electrodes, or a new anchoring mechanism on an implantable pacing lead;
- reduction in size of the wire diameter to reduce the overall pacing lead diameter;
-widening of specification limits on critical component/parameter;
-new sensors with different working principle (e.g. air bubble detector with ultrasound vs light sensor; alarm noise to light);
-change to mechanisms for preventing reflux of substances;
-removal of a design and development input.
Regarding software changes the following principles should apply (see chart C):
Non-significant change:
Examples:
-correction of an error which brings the device back to its original specification (bug fix);
-performance improvements intended to reduce latency;
-security update (e.g. cyber-security enhancements, longevity calculations);
-updates or upgrades of standard third-party operating systems, e.g. Microsoft Windows, iOS, Android;
-appearance of the user interface (e.g. new languages, layouts or graphics) provided that the changes do neither negatively affect the usability nor add new functions;
-changes to enhance the user interface without changes in performance (e.g. software change to allow the healthcare professional to select and/or change the pre- existing units of measure on a blood oxygen monitor);
-improvement to operating efficiencies (e.g. allow the queuing of multiple criteria without alteration to performance, safety or interpretation of data);
-new non-medical features, such as:
- software changes only introducing non-therapeutic and/or non-diagnostic features (e.g. printing, improved image clarity, reporting format, additional languages, barcode reader);
- change of the software manufacturer’s graphical charter (logo, colours, fonts);
- change including algorithm change impacting the control of the device without alteration of diagnosis or treatment;
- software change that disables a feature that does not interact with other features;
- displaying data on the screen which were already present in the device and part of the original conformity assessment.
Note: These examples are valid only provided that the risk/benefit ratio of the device is not negatively affected.
Significant change:
Examples:
-new or major change of operating system or any component (beyond minor changes) (e.g. Linux to Windows or iOS or Android) or new version of an operating system Windows 10 to Windows 11) if modification to the device software is required;
-new or major modification of architecture;
-change of an algorithm which impact the operating principles or impact the control of the
-device and may alter the diagnosis or therapy delivered;
-required user input replaced by closed loop algorithm;
-new user interface, such as:
- presentation of medical data in a new format or by a new dimension or measuring unit;
- keyboard input to touchscreen;
- keyboard to wireless remote control;
- software changes that impacts the way data is read or interpreted by the user, such that the treatment of the patient may be altered when compared to previous version of software;
-new medical feature or functionality that may change the diagnosis or the therapy delivered to the patient;
-new channel of interoperability (e.g. software changes that allow for wireless communication with compatible -continuous- blood glucose monitors).
Changes related to a substance or material – Chart D
Regarding changes of substances or materials the following principles should apply (see chart D):
Non-significant change:
-new or additional supplier/producer of a material within the defined specifications;
-substitution of a chemical substance in order to comply with other applicable laws and regulations e.g. REACH Regulation (EC) No 1907/2006;
-replacing a substance or material, unless considered a significant change as listed below.
Examples:
-update to material grade (e.g. additives in Loctite adhesive) with no change to specification;
-change in supplier that extrudes the polymer tubing with no change in device specifications;
-replacing a material of a sterile device’s packaging that has no function in maintaining the sterile barrier or packaging integrity;
-change in a material which serves solely as a processing aid which is not available on the finally cleaned device and therefore considered a manufacturing but not a design change;
-addition of an antioxidant to the drug coating of a combination product that reduces the degradation of the drug during production but does not alter its pharmaceutical properties or release kinetics.
Note: These examples are valid only provided that the risk/benefit ratio of the device is not negatively affected.
Significant change:
-change to a material or substance which is part of an implant and intended for direct or indirect contact with patient tissue or fluid for more than 30 days, or is part of a surgically invasive device which is absorbed;
-addition or change of a material of human/animal origin (e.g. collagen produced from skins by collagen produced from bones);
-change to a material containing a medicinal substance (23) (i.e. excipient or carrier material with influence on the delivery of the medicinal substance), or to the medicinal substance itself;
-change from a material with a low toxicological or biological risk to a material with a higher one;
-new or changed substance or material that adversely affects the safety or performance of the device and therefore negatively affects the risk/benefit ratio of the device.
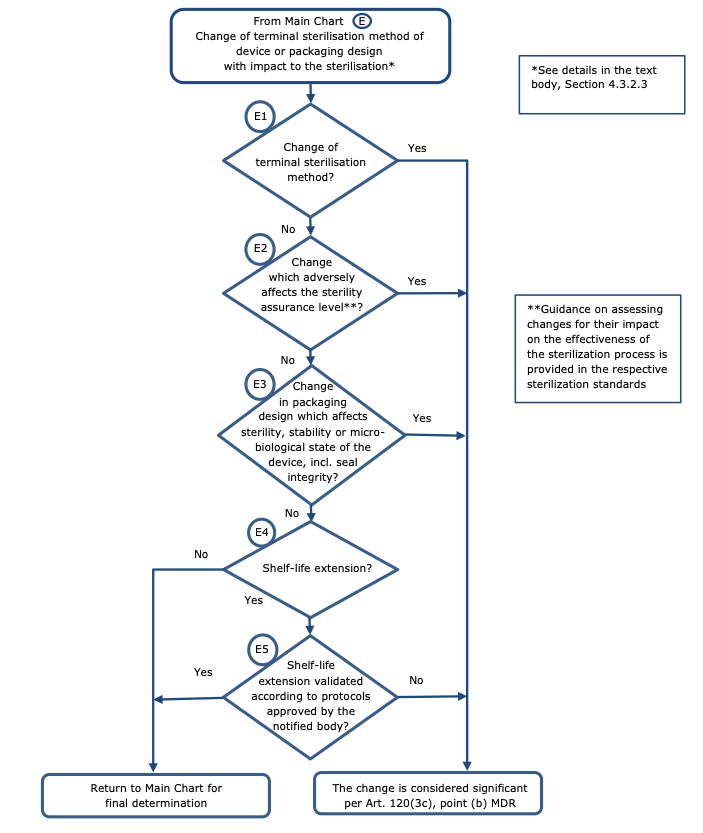
Changes related to sterilisation – Chart E
Regarding changes related to the sterilisation method or related to the design or the packaging with impact on the sterile condition of the device, the following principles should apply (see chart E):
Non-significant change:
-change of the sterilisation cycle parameters;
-extension of the shelf life validated by protocols approved by the notified body;
-new sterilisation vendor or chamber;
-change from single sterile to double sterile packaging.
Note: These examples are valid only provided that there is not impact to sterility assurance level or sterilisation residuals.
Significant change:
-change of the terminal sterilisation method (e.g. ETO to Gamma);
-change from biological indicator to parametric release;
-changing a device from labelled “non-sterile” to labelled “sterile”;
-change (e.g. of the device) which adversely affects the sterility assurance level;
-change in packaging design which affects sterility, stability or microbiological state of the device, including seal integrity;
-extension of the (labelled) shelf life which is not validated against protocols approved by the notified body.
-changes to storage or transportation conditions which could adversely affect the sterility or stability.
Annex
Design changes and changes of the intended purpose which may be considered ‘significant’ when interpreting Art. 120(3c), point (b) MDR – Main Chart
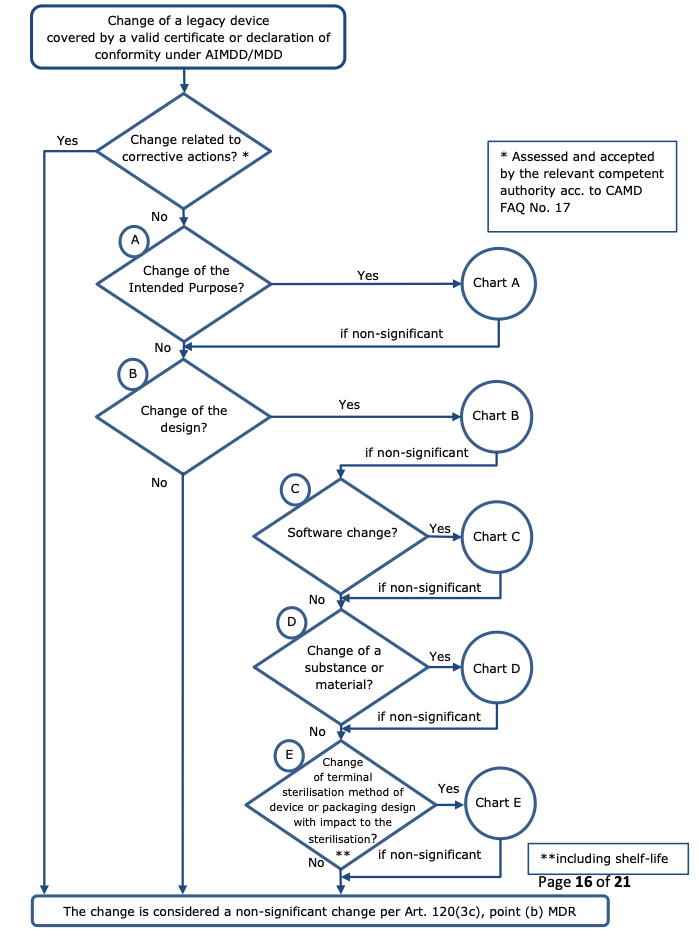
Chart A
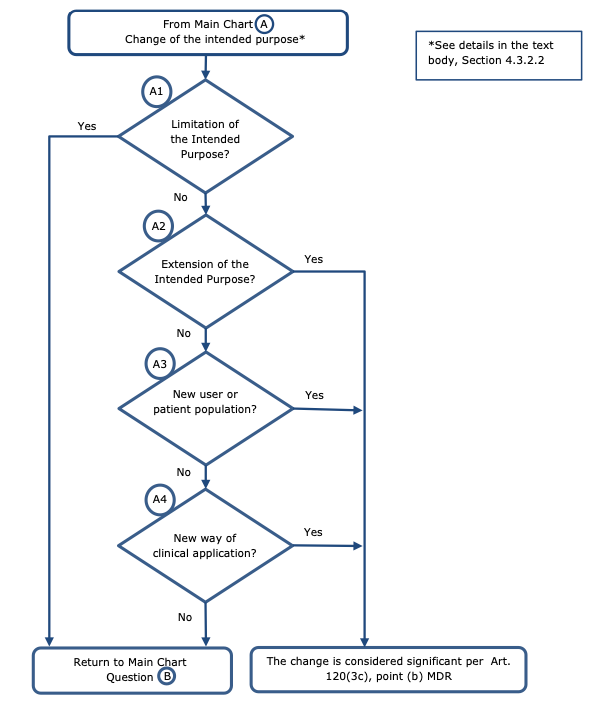
Chart B

Chart C
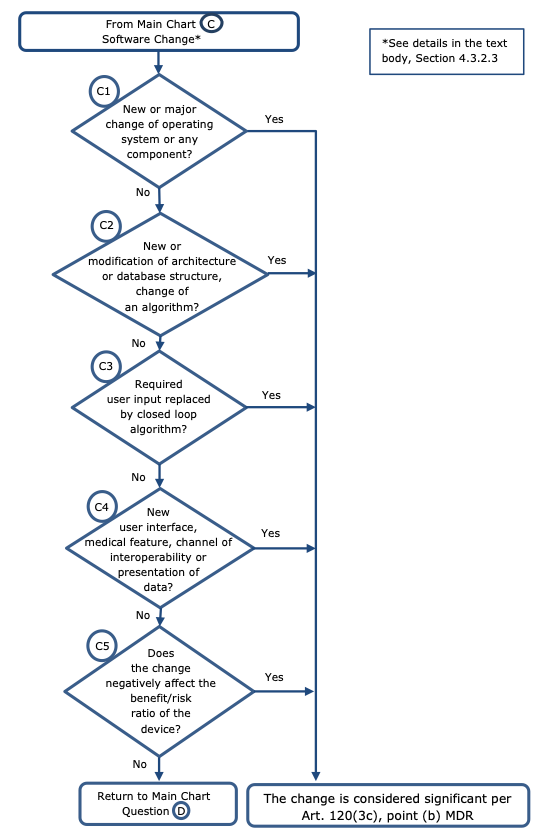
Chart D
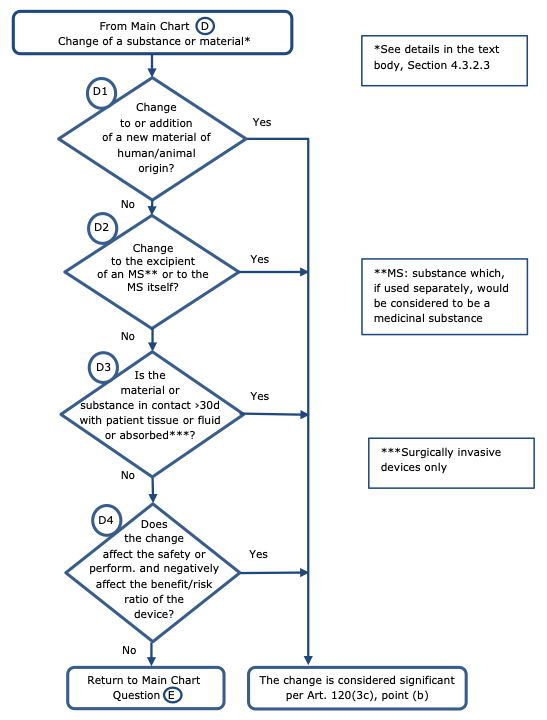
Chart E
Footnotes
(1): In case of all class III devices and class IIb implantable devices except sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors.
(2): In case of class IIb devices other than those referred to in footnote n.1, class IIa devices, and class I devices placed on the market in sterile condition or having a measuring function.
(3): MDCG 2021-25: Regulation (EU) 2017/745 – application of MDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2021 in accordance with Directives 90/385/EEC or 93/42/EEC. https://health.ec.europa.eu/system/files/2021-10/md_mdcg_2021_25_en_0.pdf
(4): See question 1 in Q&A on practical aspects related to the implementation of Regulation (EU) 2023/607,https://health.ec.europa.eu/document/download/592008f6-3456-4afb-a13a- 733a87da1b00_en?filename=mdr_proposal_extension-q-n-a_0.pdf
(5): See Article 120(3c), point (a) MDR; according to Article 120(3d) MDR, the requirements relating to post-market surveillance, market surveillance, vigilance, registration of economic operators and of devices shall apply to the devices and replace the corresponding requirements of the Directives.
(6): See Article 120(3c), point (b) MDR.
(7): See Article 120(2) MDR. This does not exclude the possibility that during the transition period an EC certificate for the manufacturer’s approved quality system issued in accordance with Annex 2/II or Annex 5/V AIMDD/MDD or Annex VI MDD, which has become invalid, is replaced by a EU QMS certificate issued in accordance with Annex IX, chapter 1, MDR, provided that the EC design examination certificate issued under Annex 2/II, section 4, AIMDD/MDD or EC type-examination certificate issued under Annex 3/III AIMDD/MDD remains valid.
(8): See Article 120(3e) MDR and MDCG 2022-4 Guidance on appropriate surveillance regarding the transitional provisions under Article 120 of the MDR with regard to devices covered by certificates according to the MDD or the AIMDD.
https://health.ec.europa.eu/system/files/2022-12/mdcg_2022-4_en.pdf [this guidance will be alinged to the amended Article 120 MDR soon].
(9): MDCG 2022-6 Guidance on significant changes regarding the transitional provision under Article 110(3) of the IVDR.https://health.ec.europa.eu/system/files/2022-05/mdcg_2022-6.pdf
(10): See Annex 2, Sect. 4.4. AIMDD and Annex II, Sect. 4.4. MDD: “The applicant shall inform the notified body … of any modification/such changes made to the approved design”; Annex 3, Sect. 6. AIMDD and Annex III, Sect. 6. MDD: “The applicant shall inform the notified body … of any modification/significant change made to the approved product”.
(11): See Annex 2, Sect. 3.4. AIMDD: “The manufacturer shall inform the notified body which has approved the quality system of any plan to alter that system”; Annex II, Sect. 3.4. MDD: “The manufacturer must inform the notified body which approved the quality system of any plan for substantial changes to the quality system or the product-range covered”; Annex V, Sect. 3.4. MDD: “The manufacturer must inform the notified body which approved the quality system of any plan for substantial changes to the quality system”.
(12): This does not mean that notified bodies are not allowed to e.g. suspend, re-insate, restrict or withdraw such certificates. Such activities should be communicated as written decisions/statements. Moreover, notified bodies should document corrections or additions to existing certificates resulting from changes that are not significant changes to design or intended purpose (e.g. administrative changes).
(13): Should the manufacturer wish to place such a changed device on the market, they may do so under the MDR.
(14): Those change notification procedures may need to be adjusted in accordance with the principles outlined in this guidance. This should be subject to notified body assessment within the surveillance activities according to Article 120(3) MDR.
(15): Provided that conditions set out in Article 120(3c) MDR are met.
(16): In case of class I devices (not certified by a NB), manufacturers may also drawn up a statement listing non- significant changes performed to the device. This statement could accompany the Declaration of conformity previously issued.
(17): This does apply only in cases where the legal entity certified under the directive(s) continues to exist. Not covered are situations where the manufacturer certified under the directive(s) will transfer device(s) covered by those directive certificate(s) to another manufacturer who will place these device(s) on the market under the MDR.
(18): Changes to a medicinal substance – including a change in its manufacturing process – which has undergone medicinal consultation should be notified to the medicines authority responsible for the consultation according to the relevant procedures (see e.g. Annex I, section 10 AIMDD or Annex I section 7.4 MDD) by the notified body.
(19): ‘Corrective action‘ refers to corrective action as defined in Article 2(67) MDR, i.e. any “action taken to eliminate the cause of a potential or actual non-conformity or undesirable situation”. This includes ‘field safety corrective actions’ (FSCA) as defined in Article 2(68) MDR. ‘Competent authority’ should generally be the authority of the Member State in which the manufacturer or its authorised representative is established. It can either be a competent authority for vigilance in accordance with Article 87 MDR or a competent authority for market surveillance in accordance with Article 93 MDR. The role of the competent authority is to assess and determine the acceptability of the (field safety) corrective action proposed by the manufacturer aimed at preventing or reducing safety risks regardless of whether the (field safety) corrective action describes a change of design or intended purpose. The assessment and acceptance of a (field safety) corrective action by a competent authority does not exempt the manufacturer from submitting changes to the relevant notified body under the AIMDD/MDD nor the notified body from assessing the change in line with the agreed arrangements (see also sections 2 and 4.1 of this guidance). The manufacturer is responsible for implementing the (field safety) corrective action, including changes in the design or intended purpose, if needed.
(20): CAMD Transition Sub Group, FAQ – MDR Transitional provisions, V1.0, 17.1.2018. https://www.camd-europe.eu/wp-content/uploads/2018/05/FAQ_MDR_180117_V1.0-1.pdf
(22): This section should be considered irrespective of whether the software is stand-alone or incorporated/utilised by/controlling a device.
(23): See footnote 18.