
MDCG 2020-16 Rev.4
Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746
Disclaimer: This document is an interactive version of the original MDCG document. We will keep it up-to-date.
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.
MDCG 2020-16 Rev.4 changes
| MDCG 2020-16 revision 4 changes | |
|---|---|
| Rule 1 – second indent | Revision of examples |
| Rule 3(m) | Footnote 8 modified |
| Rule 4(a) | Revision of examples |
| Rule 6 | Minor modifications of the rationale and revision of examples |
| Annex 1 and Annex 2 | Minor editorial changes |
| MDCG 2020-16 revision 3 changes | |
| Definitions | Addition of ‘kit’ definition |
| Rule 3(a) | Revision of examples |
| Rule 4(a) | Revised |
| Rule 5(c) | Revised |
| Rule 6 | Minor revision of example |
| MDCG 2020-16 revision 2 changes | |
| Rule 1 – second indent | Minor revision of examples |
| Rule 2 – class D examples | Editorial change |
| Rule 3 – general comment | Editorial change |
| Rule 3(f) | Revised |
| Rule 3(j) rationale | Editorial change |
| Rule 3(m) | Footnote 8 modified |
| Annex 2 | Added |
| MDCG 2020-16 revision 1 changes | |
| Rule 5(b) | Examples revised |
1. Foreword
This guidance, relating to the application of Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR) addresses the classification of in vitro diagnostic medical devices (IVDs) and provides clarifications on the classification rules as set out under Annex VIII. This classification guidance also applies to diagnostic or information society services performed on EU patients or devices put in to service through distance sales.
The guidance has been developed by an expert group representing Member State Competent Authorities, Commission services, notified bodies and industry. This guidance also takes into account the Corrigendum published in the Official Journal of the European Union on 27 December 2019. As this document is intended to provide useful information to ensure the uniform application of the relevant Regulatory provisions, it should always be read in conjunction with the IVDR.
The primary purpose of this document is to provide guidance to manufacturers, notified bodies and health institutions on how to classify an IVD prior to placing it on the market, making available on the market or putting into service in the Union. It is also intended to inform regulators and other stakeholders when assessing the class attributed to an IVD by a manufacturer or a health institution.
It is important to recall that the IVDR sets out a legal empowerment for the classification of IVDs. Therefore, and only at the request of a Member State, or on its own initiative, the Commission shall after consulting the Medical Device Coordination Group (MDCG), decide by means of implementing acts, on the application of Annex VIII to a given device, or category or group of devices, with a view to determining the classification of such devices (Article 47 (3-4) of the IVDR). To the extent necessary to resolve issues of divergent interpretation and of practical application, the Commission may adopt implementing acts, in order to ensure the uniform application of the classification rules, taking into account the relevant scientific opinions of the relevant scientific committees (Article 47 (5) of the IVDR).
Examples in this document are provided for illustrative purposes only and do not represent an exhaustive list.
Table of Contents
2. Definitions
– ‘confirmatory device’
is a device intended to be used for the confirmation of a reactive result from a first line assay.
– ‘first-line device’
is a device intended to be used to detect a marker or analyte, and which may be followed by a confirmatory assay. Devices intended to be used solely to monitor a previously determined marker or analyte are not considered first-line assays.
– ‘detecting the exposure to’ an agent
means the indirect detection of an agent (present or past exposure).
– by detecting the presence of surrogate markers, such as antibodies against components of the agent.
– ‘detecting the presence of’ an agent
means the direct detection of the agent, by detecting
– the presence of the agent itself (e.g. bacterial, viral, fungal, parasitic, protozoal agents), or
– the presence of structural components derived from the agent, such as antigens or nucleic acids.
– ‘embryo’ or ‘foetus’
an unborn refers to stages in human development after zygote formation. A zygote is considered an embryo in particular from the period of conception to approximately the eighth week, and considered a foetus following this period until birth.
Samples from the embryo or foetus include samples from the embryonic/foetal membranes, fluids and excretions, the umbilical cord, and maternal samples (e.g. blood) containing embryonic/foetal material to be examined.
– ‘offspring’
is the result of conception, at all stages of development, embryo and foetus, premature and full term neonates, child and adult.
– ‘infective/infectious agent’
is an agent capable of producing infection. This includes iatrogenic infections, i.e. those infections transmitted during medical treatment and care.
– ‘life-threatening’
are diseases, conditions or situations that in general result in death. These are often untreatable, treatment options are limited or require major medical interventions. (1)
– ‘marker’ ‘analyte’ or measurand
a substance or material; something that identifies or that is used to identify; a factor that establishes the nature of an entity or event; constituent of a sample with a measurable property.
– ‘devices for monitoring’
are used for the measurement of the analyte (measurand) levels for the purpose of adjusting treatments/interventions as required.
Devices for monitoring include the following:
– Devices which are used to assess whether an analyte remains within physiological levels or within an established therapeutic drug range. These types of devices are designed to evaluate an individual’s current state.
– Devices which are used for serial measurement, whereby multiple determinations are taken over time. These types of devices are typically used for the detection/assessment of disease progression/regression, disease recurrence, minimum residual disease, response/resistance to therapy, and/or adverse effects due to therapy. These types of devices are designed to evaluate changes in an individual’s state.
– ‘devices for screening’
are used to detect the presence of or the predisposition to a disease, disorder or other physiological state in a specimen from an individual, embryo or foetus not demonstrating clinically evident symptoms.
Depending on the nature of the condition and the targeted patient population, screening devices may be used routinely or may be restricted to „at risk“ patients.
This also includes (for example) devices intended to assess the suitability of blood, blood components, cells, tissues or organs, or in any of their derivatives for transfusion, transplantation or cell administration, with respect to transmissible agents.
– ‘devices for self testing’ (2)
means any device intended by the manufacturer to be used by lay persons, including devices used for testing services offered to lay persons by means of information society services.
– ‚kit’
means a set of components that are packaged together and intended to be used to perform a specific in vitro diagnostic examination, or a part thereof.
– ’newborn’
a newborn, or neonate, refers to an infant in the first 28 days after birth.
– ’specimen’
is a discrete portion of a body fluid or tissue taken from an individual for examination, study or analysis of one or more quantities or characteristics to determine the character of the whole. This also includes other materials, for example, hair, nails excretions, secretions, or a sample from the skin surface.
– ‘instrument’
means equipment or apparatus intended by a manufacturer to be used as an IVD medical device.
– ‘transmissible agent’
means a biological substance or entity capable of causing disease or infection in individuals through individual to individual, animal to individual, or other modes of contact.
3. Principles of Classification
In accordance with Regulation (EU) 2017/746, devices shall be classified into classes A, B, C and D, taking into account the intended purpose of the devices and their inherent risks (Article 47).
Robust risk-based classification rules are essential for the correct classification of devices according to Annex VIII as certain requirements set out by the IVDR are directly linked to device classification.
In particular, the conformity assessment route is highly dependent on classification, which is reflected in concepts such as conformity assessments scrutiny of class D devices (Article 50), the involvement of European Union reference laboratories for high-risk devices (Article 100) and a consultation with a national medicines agency or the European Medicines Agency (EMA) for companion diagnostics (Article 48(3)).
Many post-market requirements are also class dependent, e.g. annual surveillance assessments for class C and D devices, or the requirement to prepare a post-market surveillance report or Periodic Safety Update Report (PSUR) for a given device (Articles 80 and 81).
3.1 Intended purpose
The classification of a device is defined by its intended purpose, as specified by the manufacturer. This covers the use for which a device is intended according to the data supplied by the manufacturer on the label, in the instructions for use, in the performance evaluation or in promotional or sales materials or statements.
It is therefore important that the manufacturer clearly indicates the purpose for which the device is intended. Where there is a foreseeable risk that a device may be used for purposes which are covered by other classification rules and which would result in classification of a device into a higher class, a clear limitation of use should be included in the Instruction for Use (IFU) and the technical documentation of the device.
For a device to be specifically intended for a purpose referenced in a particular classification rule, the manufacturer must clearly indicate that the device is intended for such a specific purpose in the information accompanying the device. Where several classification rules or sub-rules apply, the intended purpose of the device and its claims should be sufficiently specified to enable a clear attribution of the class. Ambiguous claims may lead to higher classification (Annex VIII 1.9).
Example: A device intended to screen blood and tissue donations for syphilis would fall under class D according to rule 1. Alternatively, a device intended to diagnose syphilis, a sexually transmitted agent, in the individual would fall under class C according to rule 3a.
3.2 Application of the rules
All implementing rules, all classification rules, and all indents are to be taken into account for the classification of an IVD or an accessory for an IVD.
3.2.1 Technology
Unless otherwise specified, the rules presented below apply equally to all technologies, principles of detection or analytical procedures.
3.2.2 Specimen
Unless specified in the classification rule, the rules apply equally to all specimen types. For example, rule 3a is equally applicable to specimens derived from blood, urine, specimens collected on swabs etc. In contrast, rule 3b specifies a specimen type (cerebrospinal fluid or blood).
3.2.3 Software
Medical device software (MDSW) is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a “medical device” in the medical devices regulation or in vitro diagnostic medical devices regulation.
Software which drives a device or influences the use of the device shall fall within the same class as the device.
If the software is independent of any other device, it shall be classified in its own right (Regulation (EU) 2017/746; Annex VIII 1.4). The application of the classification rules shall be governed by the intended purpose of the MDSW.
See also: MDCG 2019-11 Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR
3.2.4 Device used in combination
When classifying devices that are used in combination, implementing rules play a key role in determining the classification of individual devices.
Examples of devices used in combination include:
- A control which is used in combination with a reagent or
- Software which drives or influences the use of an analyser.
Under the implementing rules, some devices are classified based on the classification of another device; for example:
- Software, which drives a device or influences the use of a device, shall fall within the same class as the device.
- Calibrators intended to be used with a device shall be classified in the same class as the device.
- Control materials with quantitative or qualitative assigned values intended for one specific analyte or multiple analytes shall be classified in the same class as the device.
In addition, for combinations of devices it is worth noting that implementing rules 1.1, 1.2 and 1.3 respectively state:
- Application of the classification rules shall be governed by the intended purpose of the devices.
- If a device is intended to be used in combination with another device, the classification rules shall apply separately to each of the devices.
- Accessories for an in vitro diagnostic medical device shall be classified in their own right separately from the device with which they are used.
See Annex for further examples.
4. Explanation of the IVDR Classification Rules
The following rationales and examples are intended for illustrative purposes only to assist in the use of the classification rules and are not exhaustive in their nature.
RULE 1
Devices intended to be used for the following purposes are classified as class D:
Rule 1 first indent
Devices intended to be used for the detection of the presence of, or exposure to, a transmissible agent in blood, blood components, cells, tissues or organs, or in any of their derivatives, in order to assess their suitability for transfusion, transplantation or cell administration.
RATIONALE
Rule 1; first indent applies to all devices intended to assess the suitability of blood, blood components, cells, tissues or organs or their derivatives for transfusion, transplantation or cell administration, with respect to transmissible agents. The result of the test will be a major determinant as to whether the analysed donation will be used.
Devices typically falling under this rule are intended for the detection of those agents for which the EU has harmonised the donor and donation testing requirements within the context of risk of transmission of infection (European Directives 2002/98/EC (3), 2006/17/EC (4), 2010/45/EU (corrigendum: 2010/53/EU) (5)).
Those agents are listed in the examples below.
It must be noted that:
– Member States’ national legislation may impose testing of additional transmissible agents in blood, blood components, cells, tissues or organs or their derivatives when intended for transfusion, transplantation or cell administration.
– Manufacturers may intend devices for the detection of the presence of, or exposure to, transmissible agents, other than those mentioned in the above-cited Directives, to assess the suitability of blood, blood components, cells, tissues or organs or their derivatives for transfusion, transplantation or cell administration. In that case, these devices are classified in class D according to this rule.
– Some devices can detect the same agent for completely different intended purposes than the intended purposes covered by this rule.
Where there is a foreseeable risk that tests may be misused to assess the suitability of blood, blood components, cells, tissues or organs for transfusion, transplantation or cell administration or their derivatives, a clear limitation of use should be included in the IFU and the technical documentation of the devices.
For example: “This test device is not intended to be used to detect the presence of or exposure to or for screening for transmissible agents in blood, blood components, cells, tissues, organs or any of their derivatives in order to assess their suitability for transfusion, transplantation or cell administration”.
EXAMPLES (non-exhaustive)
Devices typically falling under this rule are those that detect agents for which the EU has clearly harmonised the donor and donation testing requirements, as outlined in the relevant European directives.
European directives 2002/98/EC (on blood), and 2006/17/EC (on tissues and cells):
– Hepatitis B (HBs-Ag).
– Hepatitis C (Anti-HCV).
– Human Immunodeficiency Virus 1/2 (Anti-HIV 1/2).
In addition, the European directive 2006/17/EC (on tissues and cells) references Treponema pallidum and in certain circumstances, depending on the donor’s history and the characteristics of the tissue or cells donated: Human T-Lymphotropic Virus- I; Malaria (Plasmodium spp.); Cytomegalovirus; Trypanosoma cruzi; Epstein Barr virus; Toxoplasma gondii.
European directive 2010/45/EU (corrigendum: 2010/53/EU) (on organs) :
– Hepatitis B.
– Hepatitis C.
– Human Immunodeficiency Virus.
Member States’ national legislation may impose testing of additional transmissible agents in blood, blood components, cells, tissues or organs or their derivatives when intended for transfusion, transplantation or cell administration (for example West Nile Virus, hepatitis E virus…)
NOTE 1: devices detecting the presence of, or exposure to a transmissible agent listed above are classified in class D according to this rule only if they are intended to be used to assess the suitability for transfusion, transplantation or cell administration of blood, blood components, cells, tissues or organs, or any of their derivatives. See intended purpose.
NOTE 2: Tests intended for detection of variant Creutzfeldt-Jakob disease when available, would fall under this rule when intended to assess the suitability of blood, blood components, cells, tissues or organs or their derivatives for transfusion, transplantation or cell administration.
Rule 1 second indent
Devices intended to be used for the detection of the presence of, or exposure to, a transmissible agent that causes a life-threatening disease with a high or suspected high risk of propagation
RATIONALE
Rule 1; second indent applies to devices which are intended to be used for the detection of the presence of or the exposure to a transmissible agent that causes a life-threatening disease with a high or suspected high risk of propagation.
Several factors contribute to the risk of propagation of a pathogen within a population, namely:
– the direct or in-direct transmissibility (i.e. the probability of infection when there is contact between a susceptible and an infected individual). This includes for example consideration of the infectious dose and route of transmission e.g. aerosol, zoonosis, vector-mediated.
– the contact rate of infected and susceptible individuals (i.e. the number of contacts per time), and
– the duration of infectiousness.
For the purposes of this document, the risk of propagation of a pathogen within a population is considered to be the risk for the general European population.
This rule applies regardless of whether the test is a first line, confirmatory or supplemental assay.
EXAMPLES (non-exhaustive)
Devices intended for:
– Hepatitis B Virus.
– Hepatitis C Virus.
– Hepatitis D Virus.
– Haemorrhagic fever viruses (e.g. Ebola, Marburg, Lassa, Crimean-Congo Haemorrhagic fever.
– Human Immunodeficiency Virus 1 and 2.
– Highly virulent influenza virus.
– Human T-Lymphotropic Virus I and II.
– SARS CoV
– MERS Coronavirus.
– Smallpox virus.
– Variant Creutzfeldt-Jakob disease (when available).
NOTE 1: Variant Creutzfeldt-Jakob disease is considered under this rule due to the suspected high risk of propagation.
NOTE 2: The list of high-risk agents may be updated based on quantitative analysis of new scientific evidence on the incidence, pathogenicity, burden of mortality and morbidity, and transmission dynamics of infectious agents in the population.
Rule 1 third indent
Devices intended to be used for determining the infectious load of a life-threatening disease where monitoring is critical in the process of patient management
RATIONALE
Rule 1; third indent applies to devices intended to be used for determining the infectious load in the context of life-threatening infectious diseases, after the disease status of the patient has been previously determined, and for which patient management options, including specific treatment, are based on monitoring the infectious load.
Viral load is typically performed by nucleic acid amplification based tests (NAT).
In the case of Hepatitis B, the DNA viral load determined by molecular biology complies with the present rule due to its importance for the initiation of treatment, the evaluation of the treatment efficiency and the change of treatment, if necessary. Hepatitis B antigen tests intended for monitoring are not critical in the process of patient management and thus they do not fall under this rule.
EXAMPLES (non-exhaustive)
Devices intended to be used for determining the infectious load of:
– Hepatitis B Virus (DNA).
– Hepatitis C Virus.
– Human Immunodeficiency Virus.
RULE 2
Devices intended to be used for blood grouping
Devices intended to be used for blood grouping , or to determine foeto-maternal blood group incompatibility1, or tissue typing to ensure the immunological compatibility of blood, blood components, cells, tissue or organs that are intended for transfusion or transplantation or cell administration, are classified as class C except when intended to determine any of the following markers:
– ABO system [A (ABO1), B (ABO2), AB (ABO3)]
– Rhesus system [RH1 (D), RHW1, RH2 (C), RH3 (E), RH4 (c), RH5 (e)]
– Kell system [KEL1 (K)]
– Kidd system [JK1 (Jka), JK2 (Jkb)]
– Duffy system [FY1 (Fya), FY2 (Fyb)]
in which case they are classified as class D
1 : see Corrigendum to Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU published in Official Journal of the European Union on December 27th 2019
RATIONALE
Rule 2 applies equally to donor and recipient testing. This rule classifies blood grouping devices into two classes depending on the likelihood that a blood group marker could cause an immunogenic response or a severe haemolytic transfusion reaction. The red blood cell markers listed in this rule are critical for ensuring immunological compatibility and safe transfusion of blood and blood components. Devices related to these markers, either intended as screening, diagnostic, confirmatory or supplemental devices are class D devices.
These class D devices includes those intended for:
– The determination of the expression of ABO and Rhesus (Rh) D, Weak D, C, E, c, e in donor and recipient e.g. by serological testing or molecular genotyping.
– The determination of partial D, as these D antigen positive patients are at risk of anti-D alloimmunization.
– The detection of anti-A and anti-B antibodies for reverse ABO typing, as ABO blood grouping requires both forward (antigen) and reverse (antibody) typing.
– Screening, detection or identification of red cell antibodies for the Rh system (anti RH antibodies), Kell system (anti-KEL1 antibodies), Kidd system (anti-JK1 and anti-JK2 antibodies) and Duffy system (anti-FY1 and anti-FY2 antibodies).
– Typing of specific red blood cell antigens (KEL1, JK1, JK2 , FY1, FY2).
Devices intended for identifying markers, other than the red blood cell markers listed in this rule, which are either intended as screening, diagnostic, confirmatory or supplemental devices for blood grouping, tissue typing, or to ensure the immunological compatibility of blood, blood components, cells, tissue or organs that are intended for transfusion or transplantation or cell administration are class C devices.
All devices intended for HLA tissue typing are classified under this rule as class C devices when they are intended to be used for blood grouping, or tissue typing to ensure the immunological compatibility of blood, blood components, cells, tissue or organs that are intended for transfusion or transplantation or cell administration.
EXAMPLES (non-exhaustive)
Class D
– Device intended for molecular RhD blood group typing, targeting directly the RHD gene alleles that code for the RBC antigens, in blood donors and recipients.
– Anti-K from clone ID, Human IgM Antibody, Blood grouping reagent for transfusion purposes.
– Red blood cell kit with A1, A2, B and O cells used to detect naturally-occurring ABO blood group antibodies in patient and donor samples, in reverse grouping.
– Red blood cell kit with O red blood cells that are homozygous for Rh, Fya, Fyb, Jka and Jkb, antigens, intended to be used as antibody type and screen procedure for transfusion purposes.
– Control red blood cells, consisting of A2B R1R2 (CcD.Ee) KEL 1, intended to be used as quality control for use with ABO/Rh(D)/Kell grouping assays for transfusion purposes.
– Pre-transfusion ABO compatibility test cards intended to be used at the recipient’s bedside as precaution against ABO-incompatible purposes.
– Device used for indirect antiglobulin tests used in the screening and identification of irregular antibodies, crossmatch tests and autocontrols, according to the markers listed in rule 2 (otherwise Class C).
– Foetal RhD typing kit.
Class C
– Device intended for HLA typing by Sanger sequencing consisting of reagents for HLA-A, -B, -C, -DRB1, -DQB1 and DPB1, for transplantation purposes.
– Medical device software for high-resolution analysis of HLA sequencing data, for transplantation purposes.
– Anti-k from clone ID, Human IgG Antibody, Blood grouping reagent for transfusion purposes.
– Anti-Lea Monoclonal blood grouping reagent for transfusion purposes.
RULE 3
General comment:
Rule 3 covers a range of devices as reflected in its indents (a)-(m). Devices falling under Rule 3 (when not classified as Class D according to rules 1 & 2) are to be classified in class C, irrespective of the indent applied. It may be possible for a device to fall under more than one Rule 3 indent.
The following are in class C if they are:
(a) Devices intended for detecting the presence of, or exposure to, a sexually transmitted agent
RATIONALE
Rule 3a applies to devices detecting agents whose main mode of transmission is sexual. Sexually transmitted infections are a group of infections that may be transmitted through vaginal, oral and anal sexual intercourse. The agents that cause sexually transmitted infections may pass from person to person through blood, semen, vaginal or other bodily fluids.
EXAMPLES (non-exhaustive)
Devices intended for the detection of:
– Chlamydia trachomatis.
– Haemophilus ducreyi.
– Herpes simplex virus 1&2.
– Human papilloma virus (HPV).
– Neisseria gonorrhoeae.
– Mycoplasma hominis.
– Mycoplasma genitalium.
– Trichomonas vaginalis.
– Treponema pallidum.
– Ureaplasma urealyticum.
– Monkeypox virus.
NOTE: Although the two parasitic diseases amebiasis and giardiasis have a main mode of transmission that is nonsexual in nature (through contact with infected food or water, or faeces), these two parasites are still commonly transmitted by oro-anal sexual contact. Nevertheless, diagnostic devices intended to determine intestinal infections caused by Entamoeba histolytica and Giardia lamblia would not fall under this rule as they are captured under Rule 6.
(b) Devices intended for detecting the presence in cerebrospinal fluid or blood of an infectious agent without a high or suspected high risk of propagation
RATIONALE
Rule 3b applies to devices intended for detecting the presence of an infectious agent (either the agent itself or component thereof) e.g. bacterial, viral, fungal, parasitic, protozoal infectious agents, specifically in specimens derived from cerebrospinal fluid or blood.
Devices intended for the detection of antibodies against the infectious agent are not covered by this rule.
Several factors contribute to the risk of propagation of a pathogen within a population, namely:
– the direct or in-direct transmissibility (i.e. the probability of infection when there is contact between a susceptible and an infected individual). This includes for example consideration of the infectious dose and route of transmission e.g. aerosol, zoonosis, vector-mediated.
– the contact rate of infected and susceptible individuals (i.e. the number of contacts per time), and
– the duration of infectiousness.
For the purposes of this document, the risk of propagation of a pathogen within a population is considered to be the risk for the general European population.
This rule applies to devices independent of the route of transmission or source of the infectious agent.
This rule also applies to microbiological media intended for the detection of relevant infectious agents in cerebrospinal fluid or blood specimen.
EXAMPLES (non-exhaustive)
Devices intended for detecting the presence of:
– Bacterial pathogens: Streptococcus pneumoniae, Group B Streptococcus, Neisseria meningitidis, Haemophilus influenza type B, Listeria spp., Borrelia burgdorferi, Mycobacterium tuberculosis.
– Fungal pathogens: Cryptococcus neoformans, Aspergillus spp.
– Viral pathogens: Herpes simplex virus 1&2, human herpes virus 6, varicella zoster virus, enterovirus, West Nile virus, chikungunya, Dengue, Zika, hepatitis A, hepatitis E.
– Parasitic pathogen: Toxoplasma gondii.
– Prion agents: sporadic Creutzfeldt-Jakob disease, Gerstmann-Straussler-Scheinker Syndrome, Kuru, Fatal Familial Insomnia.
(c) Devices intended for detecting the presence of an infectious agent, if there is a significant risk that an erroneous result would cause death or severe disability to the individual, foetus or embryo being tested, or to the individual's offspring
RATIONALE
Rule 3c applies to devices intended for detecting the presence of an infectious agent (either the agent itself or component thereof) e.g. bacterial, viral, fungal, parasitic, protozoal infectious agents.
Devices intended for the detection of antibodies against the infectious agent are not covered by this rule.
This rule does not have any specimen type restrictions and is applicable to specimens being tested from the individual, foetus or embryo.
This rule applies if there is a significant risk that an erroneous result would cause death or severe disability. It is the risk of death or severe disability to an individual that must be considered. In this context, the risk of death or severe disability to the individual should take into account that an erroneous result in a healthy individual does not carry the same risk as an erroneous result in (for example) a pregnant, immunocompromised, or vulnerable individual. This rule also applies to an embryo or foetus being tested, or the individual’s offspring where an infectious agent can be detrimental to the viability/development of the embryo/foetus leading to death or disability, both current and future e.g. developmental disability
EXAMPLES (non-exhaustive)
Devices intended for detecting the presence of:
– Bacterial pathogens: Treponema pallidum, Chlamydia trachomatis, Haemophilus influenzae type B meningitis, Neisseria meningitidis, Listeria meningitis (Listeria monocytogenes), Mycobacterium leprae, Mycobacterium spp., Legionella spp., Streptococcus agalactiae, methicillin-resistant Staphylococcus aureus (MRSA) and multi-resistant Enterobacteriaceae (MRE).
– Parasitic pathogens: Toxoplasma gondii.
– Viral pathogens: Herpes simplex virus 1&2, cytomegalovirus, Rubella, Measles, Poliomyelitis, Parvovirus B19, Zika.
(d) Devices intended for pre-natal screening of women in order to determine their immune status towards transmissible agents
RATIONALE
Rule 3d applies to devices specifically intended to screen pregnant women for their immune status towards transmissible agents. These are in particular transmissible agents that may cause infections in the embryo and foetus. The term ‘immune status’ refers to the presence, absence or level of an immune response acquired by the women following an infection or vaccination.
Devices covered by this rule are intended for the screening of pregnant women before birth in order to identify the presence of an acquired appropriately targeted immune response to transmissible agents. The absence of such acquired maternal protection is associated with an enhanced risk of transmission of the agent to the embryo/foetus upon infection of the mother. These mothers may be recommended to take preventive measures.
EXAMPLES (non-exhaustive)
Devices intended to determine for prenatal screening the immune status of women towards:
– Cytomegalovirus.
– Rubella virus.
– Toxoplasma gondii.
– Varicella zoster virus.
– Zika.
– Parvovirus B19.
(e) Devices intended for determining infective disease status or immune status, where there is a risk that an erroneous result would lead to a patient management decision resulting in a life-threatening situation for the patient or for the patient's offspring
RATIONALE
Rule 3e applies to devices for both the determination of the infective disease status and the determination of the immune status of a patient.
‘Determination of infective disease status’
The determination of the infective disease status provides information on the state, condition or evolution of a disease caused by an infective agent, which may include the effectiveness of a specific treatment. In this context, the determination of the infective disease status typically involves the measurement of infective agents, antibodies to infective agents, surrogate markers or analytes in specimens from patients.
‘Determination of immune status’
The determination of the immune status provides information on the state or condition or evolution of the immune response acquired by the patient in relation to infection with a pathogenic agent, vaccinations, allergic, immunotoxic, autoimmune and alloimmune reactions such as transfusion reactions and transplant rejection reactions.
EXAMPLES (non-exhaustive)
Devices intended to determine:
– Salmonella typhi in faeces, for the assessment of the carrier-status of patients.
– Antibodies from lymphocyte secretions immunoassay intended for the detection of active Mycobacterium tuberculosis infection.
– Quantitative virus-specific NAT tests (e.g. Cytomegalovirus, John Cunningham virus, Adenovirus, Enterovirus) to monitor an immunocompromised patient’s (e.g. transplant patient) response to antiviral therapy.
– Methicillin-resistant Staphylococcus aureus and Staphylococcus aureus specific polymerase chain reaction assay for pre-surgical screening of patients to determine nasal carriage.
– Assays intended for the detection of IgM antibodies against rubella virus to identify an acute infection in pregnant women in order to determine whether specific treatment is necessary for protecting the foetus from virus-induced damage due to a lack of previously acquired immunity.
– Assays intended for the detection of IgM antibodies against HEV.
– Enzyme immunoassay intended for the quantitation of intrathecal antibodies against rubella virus in the diagnosis of rubella virus-induced encephalitis.
– Assays intended for the detection of antibodies in the recipient to potentially pathogenic viruses (e.g. anti-cytomegalovirus, anti-herpes simplex virus antibodies) to determine latent disease status of viral infection prior to organ or bone marrow transplantation.
– Screening assays comprising allergy panels, such as Multiple Allergen Simultaneous Tests (MAST), intended to detect IgE antibodies against several specific allergens that may lead to anaphylaxis, e.g. certain nutritional allergens or hymenoptera venom allergens. False-negative results with such MAST assays could increase the risk that the patient is not adequately managed for the occurrence of a life-threatening anaphylactic event.
– Assays intended for the detection of alloantibodies in the recipient associated with transplant rejection reactions, such as antibodies against – angiotensin II receptors type 1 (anti-AT1R) and against endothelin receptors type A (anti-ETAR).
– Interferon-Gamma Release Assays (IGRA) for Mycobacterium tuberculosis.
(f) Devices intended to be used as companion diagnostics
RATIONALE
Rule 3f applies to devices intended to be used as companion diagnostics.
‘Companion diagnostic’ (CDx) is defined in Article 2(7) as a device which is essential for the safe and effective use of a corresponding medicinal product to:
a) identify, before and/or during treatment, patients who are most likely to benefit from the corresponding medicinal product; or
b) identify, before and/or during treatment, patients likely to be at increased risk of serious adverse reactions as a result of treatment with the corresponding medicinal product.
For a device to be defined as a CDx, there should be a link to a medicinal product with an International Non-proprietary Name (INN) (6).
The identification of patients may comprise a quantitative or qualitative determination of specific markers. Such specific markers can be present in healthy subjects and/or in patients.
The emphasis ‘before and/or during treatment’ implies that CDxs may be intended to be applied before a treatment with a corresponding medicinal product is initiated, or during treatment, to identify if (still) the patient is (a) likely to benefit from the corresponding medicinal product or (b) likely to be at increased risk of serious adverse reactions.
Devices that are intended to be used for monitoring treatment with a medicinal product in order to ensure that the concentration of relevant substances in the human body is within the therapeutic window are not considered to be CDxs (e.g. devices intended for blood glucose monitoring, devices intended for measurement of cyclosporine concentration in blood, devices intended for measurement of metabolites of a medicinal product).
Devices intended to determine quantitative or qualitative specific marker(s) to establish the dosage of a particular medicinal product, for patients that are already eligible to receive that medicinal product, are not considered to be CDxs. For example, devices intended to measure creatinine concentration can be used for estimating kidney function to determine the optimal dosage of medicinal products with renal elimination. Another example is devices identifying CYP2D6 or CYP2C19 genotypes of a patient to determine the appropriate dosage of an already prescribed medication.
Annex II to this guidance provides a flowchart to help determine whether an IVD is a CDx.
EXAMPLES (non-exhaustive)
General CDx examples (valid provided that the device is essential for the safe and effective use of the corresponding medicinal product):
– A device intended to identify a genotype, single or multiple genetic and/or genomic variants
– A device intended to identify a marker (receptor, transporter, other protein-based biomarker or its variant) specifically targeted by the corresponding medicinal product.
– Devices intended to detect antibodies against a specific medicinal product during the course of treatment.
– Devices intended to identify patients who are expected to benefit from treatment with a specific medicinal product, based on the absence of a marker.
Specific CDx examples:
– A device intended for the qualitative detection of anaplastic lymphoma kinase (ALK) protein in formalin-fixed, paraffin-embedded (FFPE) non-small cell lung carcinoma (NSCLC) tissue, intended as an aid in identifying patients eligible for treatment with crizotinib or ceritinib.
– A device intended for the quantitative detection of BCR-ABL1 transcripts and the ABL1 endogenous control mRNA in peripheral blood specimens from patients previously diagnosed with t(9:22) positive chronic myeloid leukemia, during treatment with nilotinib.
– A qualitative immunohistochemical device using monoclonal mouse Anti-PD-L1, intended for use in the detection of PD-L1 protein in FFPE NSCLC and gastric or gastroesophageal junction (GEJ) adenocarcinoma tissues, that is indicated as an aid in identifying patients for treatment with pembrolizumab.
– A polymerase chain reaction based device for the qualitative detection of isocitrate dehydrogenase-2 (IDH2) gene point mutations in DNA extracted from human blood or bone marrow, that is indicated as an aid in identifying acute myeloid leukemia patients with mutated IDH2 enzymes for treatment with enasidenib.
– A device intended for the demonstration of individuals homozygous for the non-functional DPYD variant DPYD*2A, that typically have complete dihydropyrimidine dehydrogenase (DPD) deficiency. The DPYD gene encodes DPD, an enzyme that catalyzes the rate-limiting step in fluorouracil metabolism. Capecitabine, a chemotherapy agent used in the treatment of colon cancer, metastatic colorectal cancer, and metastatic breast cancer, is a prodrug that is enzymatically converted to its active form, fluorouracil. Individuals who are carriers of non-functional DPYD variants, may not be able to metabolise capecitabine at normal rates, and are at risk of potentially life-threatening capecitabine toxicity, such as bone marrow suppression and neurotoxicity.
– A device intended to identify defined EGFR mutations in order to administer the tyrosine-kinase inhibitor dacomitinib for the treatment of adult patients with locally advanced or metastatic (NSCLC) and EGFR-activating mutations.
– A next-generation sequencing (NGS) based device to evaluate KRAS/NRAS genetic variants to determine the presence of mutations affecting the efficacy of vectibix for treatment of metastatic colorectal cancer.
– A device intended for the detection of 7 somatic mutations in the human KRAS oncogene to aid in the identification of colorectal cancer patients for treatment with cetuximab of with panitumumab based on a KRAS no mutation detected test result.
(g) Devices intended to be used for disease staging, where there is a risk that an erroneous result would lead to a patient management decision resulting in a life-threatening situation for the patient or for the patient's offspring
RATIONALE
Rule 3g applies to devices where the intended purpose is for the staging of a disease.
Disease staging involves the determination of distinct phases or periods in the course of a disease, or the level of severity of a disease that can be assessed by, for example, the life history of an individual, organism, markers or any biological or physiological process. The purpose of disease staging is to provide information with respect to patient management, the appropriateness and accuracy of treatment decisions, and/or for prognosis or prediction.
For the majority of diseases, determination of disease stage involves taking into account the complete clinical picture which may involve investigative procedures, examining the patient/patient history and measurement of markers or analytes in patient specimens.
This rule applies to devices where the information provided from the device is intended to be used for disease staging and where the patient management decision, being solely or principally based on an erroneous result, has the potential to result in a life-threatening situation.
This rule does not include:
– Devices for the staging of cancer as these devices are classified under rule 3h.
– Devices where the result is not intended to be used for staging a particular disease e.g. markers which are indicative of the presence of an affected health condition in general.
EXAMPLES (non-exhaustive)
– Device intended for the quantitative measurement of Brain type natriuretic peptide (BNP) in whole blood or plasma samples, for the assessment of the severity of congestive heart failure.
– Devices intended for staging of enhanced liver fibrosis (ELF) for detecting the following markers: hyaluronic acid, procollagen III amino terminal peptide, tissue inhibitor or metalloproteinase.
– Medical device software intended to generate an estimated glomerular filtration rate (eGFR) or albumin creatinine ratio (ACR) for staging acute kidney injury (AKI).
– Medical device software intended to generate an enhanced liver fibrosis (ELF) score which correlates to the level of fibrosis.
– Medical device software intended to generate a model for end stage liver disease (MELD) score.
(h) Devices intended to be used in screening, diagnosis, or staging of cancer
RATIONALE
Rule 3h applies to devices with the specific intended purpose to be used in screening, diagnosis or staging of cancer.
‘Cancer’ is the uncontrolled growth and spread of cells. It can affect almost any part of the body. The growths often invade surrounding tissue and can metastasise to distant sites. Cancer is a generic term for a large group of diseases characterised by the growth of abnormal cells which can invade nearby tissues and may spread to other parts of the body through the blood and lymph systems. Other common terms used are ‘malignant tumours’ and ‘malignant neoplasms’.
The scope of this rule is limited to cancer. Two distinct cases should be considered:
1. Cancer is a term for diseases (including malignant neoplasia or malignant tumours) characterised by abnormal cells which divide without control and invade nearby tissues. Additionally, these cells can also spread to other parts of the body through the blood and lymph systems, in the process that leads to the development of secondary tumours or metastases. Many cancers form solid tumours, which are masses of tissue. Cancers of the blood, such as leukaemia, generally do not form solid tumours.
Cells may present hyperplasia (increased density of cells) and dysplasia (abnormal appearance) or form a carcinoma in situ (no invasion of nearby tissues). These precancerous or premalignant cells may or may not develop into cancer.
This rule applies to devices for both cancerous and precancerous conditions, where the devices are intended to be used in screening, diagnosis, or staging of cancer.
Disease staging involves the determination of distinct phases or periods in the course of a disease, or level of severity of a disease that can be assessed by, for example, the life history of an individual, organism, markers or any biological or physiological process. The purpose of disease staging is to provide information with respect to patient management, the appropriateness and accuracy of treatment decisions, and/or for prognosis or prediction.
Non-malignant neoplasms or non-malignant tumours do not spread into, or invade, nearby tissues. Therefore, they do not fulfil the criteria of cancer. These benign tumours may grow larger, and the growth tends to be slow.
Devices that are intended to be used in screening, diagnosis, or staging of cancer, may have the following functions: screening, patient management, monitoring, diagnosis or aid to diagnosis, prognosis and prediction.
EXAMPLES (non-exhaustive)
– A faecal occult blood screening test (FOBT) or faecal immunochemical test (FIT) specifically intended to be used in colon cancer screening.
– A device intended for the quantitative/qualitative determination of IgG antibodies to Helicobacter pylori in human blood samples specifically intended to be used in gastric cancer screening.
– Papanicolaou (Pap) stain automated cervical cytology screening system, intended to process Pap cervical cytology slides and classify the cervical specimen as either normal or abnormal.
– A qualitative real-time PCR test intended for the detection of high-risk genotypes of Human Papillomaviruses for use in cervical cancer screening.
– Immunohistochemistry assay intended for the detection of c-KIT or CD117 tyrosine kinase receptor expression in normal and neoplastic formalin-fixed, paraffin-embedded tissues for histological evaluation, and gene mutation testing for KIT and platelet-derived growth factor receptor alpha in (familial) gastro-intestinal stromal tumor.
– Assay for the quantitative determination of the cancer associated antigen CA 125 (celomic epithelium-related glycoprotein associated with epithelial ovarian cancer) in serum.
– Immunohistochemistry assay intended to detect progesterone receptor in breast tumours to be used as an aid in the management, prognosis, and prediction of therapy outcome of breast carcinoma.
– Fluorescence in situ hybridisation (FISH) panels intended for the diagnosis of e.g. lymphoma, multiple myeloma and leukaemia.
– Targeted next generation sequencing test intended to be used in (haemato)-oncology, to detect acquired somatic mutations in DNA isolated from formalin-fixed paraffin embedded (FFPE) tumour tissue specimens.
– BRCA1 device intended for the detection of deletions or duplications in the human BRCA1 gene in order to confirm a potential cause and clinical diagnosis for hereditary breast and ovarian cancer and for molecular genetic testing of at-risk family members.
– Device applied in testing services intended for the analysis of 35 genes relevant to digestive tract tumours (various forms of colorectal cancer, stomach cancer and pancreatic cancer), breast cancer, ovarian cancer, skin cancer, thyroid tumours, and endocrine tumours (panel), intended to provide information on whether an individual carries genetic alterations that favour the onset of specific tumour diseases, identifying these genetic predispositions.
– Circulating Tumour Cell Kit (Epithelial) intended for the enumeration of circulating tumour cells (CTC) of epithelial origin in whole blood. The test is to be used as an aid in the monitoring of patients with metastatic breast, colorectal or prostate cancer. Serial testing for CTC should be used in conjunction with other clinical methods for monitoring metastatic breast, colorectal and prostate cancer, to allow assessment of patient prognosis and is predictive of progression free survival and overall survival.
– Breast carcinoma cell line (SK-BR-3) CTC Cell Control Kit intended as an assay control to ensure that the sample detection and identification systems are performing when using the CTC Kit. They express epithelial cell markers recognised by the antibodies in the Circulating Tumour Cell Kit and are used as a control for the performance of the assay.
– An image analysis medical device software intended to aid in the detection and semi-quantitative measurement of programmed death ligand 1 (PD-L1) protein in FFPE lung tissue. The algorithm is an adjunctive computer-assisted methodology for a qualified pathologist in the acquisition and measurement of images from microscope glass slides of FFPE patient lung tissue stained for the presence of PD-L1 protein using an associated PD-L1 Assay. It scans digitised images of tissue specimens stained with the associated PD-L1 assay and provides raw numbers (scores) for immune cells and tumour cells counts in the specimen, along with a percent positivity score.
(i) Devices intended for human genetic testing
RATIONALE
Rule 3i applies to devices for human genetic testing. Genetic testing involves the detection of specific alleles, mutations, genotypes, karyotypes or epigenetic changes that are associated with heritable traits, diseases or predispositions to disease for the individual or their descendants.
Several methods can be used for genetic testing (for example) (7):
– Molecular genetic tests (or gene tests) study single genes or short lengths of DNA to identify its constitution, or variations or mutations that lead to a genetic disorder.
– Chromosomal genetic tests analyse whole chromosomes or long lengths of DNA to see if there are large genetic changes, such as an extra copy of a chromosome which causes a genetic condition.
The results of a genetic test can provide a medical status, confirm or rule out a suspected genetic condition or determine a person’s chance of developing or passing on a genetic disorder.
Some examples include devices intended for:
– Newborn Screening: Newborn screening is used just after birth to identify genetic disorders, to detect potentially fatal or disabling conditions. Such early detection allows treatment to begin immediately, which can reduce or eliminate the effects of the condition.
– Diagnostic testing: Diagnostic testing is used to identify or rule out a specific genetic or chromosomal condition.
– Carrier testing: Carrier testing is used to identify people who carry one copy of a gene mutation that could result in a genetic disorder in one’s offspring. For some genetic disorders, two copies of the gene mutation are required to cause the genetic disorder (autosomal recessive). Whereas for others, one copy of the gene mutation is required either i) in the absence of a second normal copy resulting in the genetic disorder (X-Linked recessive) or ii) in the presence of a normal copy can result in a genetic disorder (autosomal dominant). This type of testing provides information about a couple’s risk of having a child with a genetic condition.
– Prenatal testing: Prenatal testing is used to detect changes in a foetus’s genes or chromosomes before birth.
– Preimplantation testing: Preimplantation testing, also called preimplantation genetic diagnosis (PGD), is a specialised technique used to detect genetic changes in embryos obtained through in vitro fertilisation.
– Predictive and presymptomatic testing: Predictive and presymptomatic types of testing are used to detect gene mutations associated with disorders that appear after birth, often later in life. Predictive testing can identify mutations that increase a person’s risk of developing disorders with a genetic basis. Presymptomatic testing can determine whether a person will develop a late-onset genetic disorder.
– Direct-to-Consumer (DTC) genetic testing: genetic testing provided through advertising and selling or (free) provision of genetic tests directly to consumers.
EXAMPLES (non-exhaustive)
Genetic testing may include devices intended to detect:
– Chromosomal conditions e.g. trisomy 21, trisomy 18, XXX syndrome.
– Abnormalities in genes associated with thrombophilia e.g. genes which code for factor V and prothrombin.
– Hereditary cancer syndromes e.g. hereditary breast/ovarian cancer (BRCA1/BRCA2 genes).
– Genetic risk Factors e.g. rheumatoid arthritis HLA DRB1, ankylosing spondylitis HLA B27, osteo-arthritis, pre-senilin mutation.
– Monogenetic disorders e.g. hemochromatosis, Huntington’s disease, Tay Sacs, cystic fibrosis.
– Pharmacogenomic tests e.g. CYP liver enzymes CYP2C9 and CYP2C19.
– Preimplantation genetic diagnosis.
– XY disorders e.g. haemophilia, Duchenne muscular dystrophy, Fragile X.
(j) Devices intended for monitoring of levels of medicinal products, substances or biological components, when there is a risk that an erroneous result will lead to a patient management decision resulting in a life-threatening situation for the patient or for the patient's offspring
RATIONALE
Rule 3j applies to devices intended to monitor an analyte with the purpose of adjusting patient management, such as treatments/interventions, as required i.e. it is intended to be used for observing, checking, or keeping a record of the level, activity, presence, absence etc. of an analyte.
Monitoring tests may be intended to evaluate an individual’s current state and/or changes in an individual’s state. This is likely to be achieved by repeated or multiple determinations of an analyte over time, at appropriate intervals. These devices are intended to determine whether results are within expectation, for the detection/assessment of disease progression/regression, disease recurrence, minimum residual disease, response/resistance to therapy, and/or adverse effects due to therapy.
This rule does not apply to devices intended to be used in the diagnosis or screening of a condition where only a single measurement is required for this purpose, but would apply to diagnostic tests where multiple/serial measurements over time are intended by the manufacturer and where an erroneous result may result in a life threatening situation for the patient or their offspring e.g. when monitoring the change in concentration of a biological compound over time to aid diagnosis, such as with troponin to help determine an acute cardiac event.
Rule 3j is also applicable to the monitoring of non-life threatening conditions. It covers a wide range of analytes where the device provides an important, critical, or sole determinant for the correct patient management decision and an erroneous result may result in the life of the patient or patient’s offspring being at risk due to inappropriate treatment decisions.
Analytes measured by devices intended for monitoring may be medicinal products (Article 1(2) of Directive 2001/83/EC, as amended), substances (drug, chemical, or biological entity/component) or biological components (pertaining to living organisms, or components of a living organism – this would include for example: antibodies, endogenous markers, platelets, cord blood, bone marrow, stem cells etc).
If the device is intended for a specific intended target population (e.g. paediatrics, pregnant women, immunocompromised individuals, etc.) then the risk to this population should be taken into account when determining if there is a risk that an erroneous result would lead to a patient management decision resulting in a life-threatening situation for the patient or their offspring.
With respect to the patient’s offspring, the viability/development of the embryo/foetus, both current and future shall be taken into account.
EXAMPLES (non-exhaustive)
Devices intended for monitoring:
– Cardiac marker for acute presenting patients: Troponin I, Troponin T, CKMB (when intended for monitoring cardiac muscle injury).
– Cortisol levels monitoring e.g. for patients with cortisol insufficiency.
– PT/APTT when used to assess major bleeds in acute presentations or patients with acute coagulopathy or for coumadin monitoring in patients without diagnosed coagulation disorder.
– Lithium for patients being treated for bipolar disorders.
– Methotrexate when used for treating non-life threatening conditions such as vasculitis, rheumatoid arthritis and psoriatic arthritis).
– Immunosuppressive (anti-rejection) medicinal products e.g. cyclosporine, sirolimus, tacrolimus.
– Antibiotic where under/over treatment can have a serious impact on individual or offspring e.g gentamicin.
– Anti-RhD antibody levels in pregnant women given additional Anti-D.
– Blood amylase e.g. acute pancreatitis, perforated peptic ulcer, acute biliary obstruction.
– Acute phase reactants e.g. C- reactive protein (CRP), procalcitonin when intended to be used to monitor infection response to therapy for life threatening conditions such as sepsis, necrotizing skin or tissue conditions, infective endocarditis, bacterial meningitis etc.
– Full blood count when used for monitoring for the development of a life threatening haematological disorder in patients being treated for other disorders/conditions, where this risk exists e.g. monitoring of patients with a diagnosis of schizophrenia for neutropenia/agranulocytosis.
– Bilirubin in response to treatment of neonatal jaundice.
(k) Devices intended for management of patients suffering from a life-threatening disease or condition
RATIONALE
Rule 3k applies to devices intended for patients diagnosed with life-threatening diseases or conditions.
The device provides an important, critical, or sole determinant for the correct patient management decision, and provides information for the purpose of patient management, such as treatments/interventions, as required.
The classification of these devices is primarily based on the life-threatening nature of the disease or condition and the impact of the provided information on patient management (e.g. determining an initial course of therapy or erroneous decision resulting in life-threatening harm to the patient). This includes devices intended to detect drug resistant pathogens associated with a life-threatening condition (e.g. sepsis, necrosing skin or tissue condition) directly from collected specimen such as blood, skin or tissues, in order to take a patient management decision. However, rule 3k does not apply to devices used in conjunction with microbiological culture methods that are only intended to test drug resistance of an already detected pathogen including drug sensitivity testing such as sensitivity discs and tablets or Minimum Inhibitory Concentration (MIC) panels, where such devices are not intended for the management of patients suffering from a life-threatening infection.
EXAMPLES (non-exhaustive)
Devices intended for:
– Enumeration of CD4 T lymphocytes in HIV infected patients to initiate treatment and ascertain the anti-viral therapy response.
– Measurement of D-Dimers in patients with thrombotic disorders.
– Laboratory risk score calculator indicator for necrotising fasciitis in necrotising soft tissue infections.
– HbA1c and blood glucose tests for the management of patients with diabetes.
– Monitoring anticoagulant therapy e.g. prothrombin Time/INR (warfarin), APTT (unfractionated heparin), anti-Xa chromogenic assays (low molecular weight heparin (LMWH), fondaparinux, rivaroxaban, and apixaban), anti-IIa chromogenic and clot-based assays (argatroban, bivalirudin, hirudin, and dabigatran).
– Digoxin monitoring.
– Anti-retroviral resistance testing in HIV infected patients.
(l) Devices intended for screening for congenital disorders in the embryo or foetus
RATIONALE
Rule 3l applies to devices for routine screening of embryo/foetus, and also specific screening for embryo/foetus whose families have known inherited conditions or where specific populations are at greater risk of an inherited condition e.g. Sickle cell.
Rule 3i also applies to preimplantation and genetic screening tests.
EXAMPLES (non-exhaustive)
– Devices intended for screening of foetal aneuploidies (e.g. trisomy 13, trisomy 18 and trisomy 21), which include devices intended for the measurement of biochemical maternal serum markers.
– Reagents and medical device software evaluating the risk of foetal aneuploidies based on biochemical markers and other information, in particular non-invasive prenatal tests (NIPT).
– Devices intended to determine the foetal sex in cell-free foetal DNA in maternal blood, in the remit of sex-depending congenital disorders.
– Genetic test for cystic fibrosis.
– Genetic test for sickle cell disease.
– Huntington’s chorea.
– Tay Sachs.
– Thalassaemia and other haemoglobin disorders.
(m) Devices intended for screening for congenital disorders in new-born babies where failure to detect and treat such disorders could lead to life-threatening situations or severe disabilities
RATIONALE
Rule 3m applies to devices intended for screening new-born babies for a defect which is present from birth i.e. a structural or functional abnormalities, including metabolic disorders, where an erroneous result could lead to a failure to detect and treat such birth disorders, which could lead to a life-threatening situation or severe disability of the individual. This includes genetic testing where the intended purpose covers screening for congenital disorders in neonates.
This rule applies to devices intended to be used for screening (8) new-born babies shortly after birth for disorders that are treatable, but not clinically evident in the new-born period. Some of the conditions included in new-born screening are only detectable after irreversible damage has been done.
New-born babies who screen positive undergo further testing to confirm whether they are affected with a congenital disorder. For these confirmatory and supplemental devices, and for congenital disorders that are clinically evident, in particular rules 3j and 3k shall be taken into consideration.
EXAMPLES (non-exhaustive)
Examples of devices intended for screening in new-born babies for congenital disorders:
– Beta-thalassaemia.
– Biotinidase deficiency.
– Congenital adrenal hyperplasia – e.g 17-hydroxyprogesterone (17-OHP).
– Congenital hypothyroidism – e.g thyroxine.
– Cystic fibrosis – e.g. mutation and variant screening, immunoreactive trypsin.
– Galactosaemia – e.g. total galactose or galactose-1-phosphate uridyltransferase.
– Glutaric aciduria type 1.
– Hyperphenylalaninaemia / phenylketonuria e.g phenylalanine (in blood); phenylpyruvic, phenyllactic, 2-OH phenylacetic (in urine).
– Homocystineuria (pyridoxine unresponsive) e.g. free homocystine, total homocysteine, and methionine (in blood and urine).
– Isovaleric acidaemia.
– Maple syrup disease (MSUD IA, IB, II) – e.g. branched-chain amino acids, allo isoleucine (in blood); branched-chain 2-ketoacids, branched-chain 2- hydroxy acids (in urine).
– Medium-chain acyl-CoA dehydrogenase deficiency – e.g. acylcarnitine measurement.
– Methylmalonic aciduria including cblA, cblB, cblC and cblD.
– Propionic aciduria.
– N-Acetylglutamate synthase deficiency – e.g. glutamine, alanine, citrulline, arginine (in blood).
– Sickle-cell disease.
– Tyrosinemia (I, II, III) – e.g. tyrosine (in blood); succinylacetone, 4-OH phenylpyruvic, 4-OH phenyllactic acids (in urine).
– Severe combined immunodeficiency (SCID) e.g. by TREC/KREC determination.
RULE 4
(a) Devices intended for self-testing are classified as class C, except for devices for the detection of pregnancy, for fertility testing and for determining cholesterol level, and devices for the detection of glucose, erythrocytes, leucocytes and bacteria in urine, which are classified as class B
RATIONALE
Rule 4a applies to devices intended for self-testing. All devices intended for self-testing are classified as class C, except:
– devices for the detection of pregnancy, for fertility testing, and for determining cholesterol level in any specimen, and
– devices for the detection of glucose, erythrocytes, leucocytes and bacteria in urine,
which are classified as class B.
If a device simultaneously detects a marker that falls under this rule as a class C in addition to a marker listed as an exception (class B), then the device is
classified as class C according to the implementing rules 1.8 and 1.9.
– those classified as class D according to the implementing rules 1.8 and 1.9.
Devices intended by the manufacturer to be used for testing services offered to lay persons, including those offered by means of information society services (9), are considered devices for self-testing when the lay persons carry out at least a part of the testing procedure, such as adding a reagent or placing the specimen on a test cassette. Such actions do not include those needed to collect the specimen or to ensure specimen integrity and stability (see Rule 5).
Standalone specimen receptacles and kits intended for specimen collection, when intended to be used by lay persons for specimen collection only, including those offered to lay persons by means of information society services, are not considered devices for self-testing (see Rule 5).
EXAMPLES (non-exhaustive)
– Meters and strips (containing integrated testing reagent) for self-testing of capillary blood glucose are in class C.
– Self-testing devices for blood clotting, e.g. measurement of International Normalised Ratio (INR) are in class C.
– Devices intended to measure the levels of calprotectin where the lay person collects the stool specimen, carries out the testing procedure using the test cassette and sends an image of the result to be interpreted by a healthcare professional are in class C.
– Self-testing devices for detection of HIV antibodies from a fingerprick blood sample are in class D (as per Rule 1 and implementing rule 1.9).
– Self-testing devices intended for the detection of SARS CoV-2 or antibodies against SARS CoV-2 are in class C.
(b) Devices intended for near-patient testing are classified in their own right
RATIONALE
Rule 4b applies to devices intended for near-patient testing. The classification of devices for near-patient testing follows the intended purpose of the device, as established by the manufacturer. This brings the classification of devices for near-patient testing in line with that of other devices intended for professional use. The manufacturer should check all the rules to determine the correct device classification.
EXAMPLES (non-exhaustive)
For the below examples, the device is intended for near-patient testing:
– Class D (under Rule 1): Rapid test for detection of human immunodeficiency virus.
– Class D (under Rule 2): Pre-transfusion ABO compatibility test cards intended to be used at the recipients‘ bedside as precaution against ABO- incompatible transfusion.
– Class C (under Rule 3): Blood glucose reagents / strips for patient monitoring.
– Class C (under Rule 3): Mobile cardiac marker monitoring test for acute presenting patients: Troponin I, Troponin T, CKMB (when intended to be used for monitoring cardiac muscle injury).
– Class C (under Rule 3): Rapid test for the detection of methicillin-resistant Staphylococcus aureus.
– Class B (under Rule 6): Urine dipstick to determine urinary tract infection at point of care.
– Class B (under Rule 6): Quantitative test for haemoglobin as an aid in diagnosing iron deficiency.
– Class B (See Rule 6): Rapid tests for the detection of Group A Strep, Respiratory Syncytial Virus, and Influenza virus(es).
RULE 5
(a) Products for general laboratory use, accessories which possess no critical characteristics, buffer solutions, washing solutions, and general culture media and histological stains, intended by the manufacturer to make them suitable for in vitro diagnostic procedures relating to a specific examination
RATIONALE
Rule 5a applies to general laboratory products like pipettes, stain powders, glass microscope slides, centrifuges, pipette tips or instrument liquid collection containers, buffers which usually do not fall under the definition of an IVD medical device. However, as specified in Regulation (EU) 2017/746 Article 1 (3a)
’This regulation does not apply to (a) products for general laboratory use (…), unless such products, in view (…) are specifically intended by their manufacturer to be used for in vitro diagnostic examinations.’
As a consequence, if such products are specifically intended by the manufacturer to be used for in vitro diagnostic examinations, then they are considered as IVDs and are captured by rule 5.
‚Accessory for an in vitro diagnostic medical device‘ as defined under Regulation (EU) 2017/746 article 2 (4), ‘means an article which, whilst not being itself an in vitro diagnostic medical device, is intended by its manufacturer to be used together with one or several particular in vitro diagnostic medical device(s) to specifically enable the in vitro diagnostic medical device(s) to be used in accordance with its/their intended purpose(s) or to specifically and directly assist the medical functionality of the in vitro diagnostic medical device(s) in terms of its/their intended purpose(s)’.
Whilst not being an IVD in themselves, accessories are to be used in conjunction with a specific IVD. They possess one or more specific characteristics to specifically enable an IVD to be used in accordance with its intended purpose or to assist the medical functionality of the IVD. Accessories are mentioned in rule 5 (a) in combination with the attribute ‘accessories which possess no critical characteristics’. This emphasizes that such products can negatively influence the benefit-risk ratio of the entire in vitro diagnostic medical device.
EXAMPLES (non-exhaustive)
– General microbiological culture media containing selecting agents, antimicrobial chromogenic agents, chemical indicators for colour differentiation.
– Solutions like cleaners, buffer solutions, lysing solutions, diluents specified for use with an IVD.
– Pipette with a specific fixed one volume specifically intended for a particular IVD test with specified human sample, e.g. blood coagulation pipettes with automatic timing (Accessory of coagulometer).
– General staining reagents like hematoxylin, eosin, pap and grams iodine.
– Kits for Isolation and purification of nucleic acids from human specimens.
– Library Prep reagents for preparation of DNA for downstream analysis by NGS sequencing.
– Nucleic acid quantitation kits.
– General reagents (not assay specific) used with a Class A instrument, e.g. general sequencing consumable reagents used with a sequencer.
(b) Instruments intended by the manufacturer specifically to be used for in vitro diagnostic procedures
RATIONALE
Rule 5b applies to instruments specifically intended by the manufacturer for in vitro diagnostic procedures. These instruments are classified as class A, whereas reagents and kits are classified in their own right.
Due to their interdependence, the performance of the reagent on this instrument will be part of the conformity assessment of the reagent. If the instrument has an independent measuring function which does not use any additional reagents, it is classified according to the intended purpose of the analysis (including instruments controls or instrument quality control). e.g. cell counting analysers used in haematology, ion selective electrodes, instruments measuring blood gases or glucose via its sensors, specific gravity measurements in urine analysis, mass spectrophotometer for bacteria identification, erythrocyte sedimentation rate analyser etc.
EXAMPLES (non-exhaustive)
– Enzyme immunoassay analyser, PCR thermocycler, sequencer for NGS applications, clinical chemistry analyser.
– Instrument for automated purification of nucleic acids and PCR set-up.
(c) Specimen receptacles
RATIONALE
Specimen receptacles are specimen containers or evacuated or non-evacuated tubes, empty or prefilled with a fixative solution or other general reagent intended for primary containment, preservation, transport and storage of biological specimens (e.g. cells, tissues specimens, urine, faeces) for the purpose of in vitro diagnostic examinations. They are classified in class A.
In line with their definition in Article 2(3) of the IVDR, specimen receptacles are intended for the primary containment and preservation of specimens, i.e. they cannot have any integrated testing functions.
Specimen receptacles may be placed on the market as standalone devices, but also as part of a kit intended for specimen collection or as part of a kit intended for testing.
Kits intended for specimen collection must include at least one IVD (specimen receptacle). The kit may also include devices other than IVDs, such as a medical device or components which are covered neither by the IVDR nor by the MDR.
The use of specimen receptacles or kits intended for specimen collection may require an action performed by the user to collect the specimen, such as mouth wash or gargling, or an action on the specimen to ensure specimen integrity and stability, according to the IFU, such as pipetting or buffer addition (i.e. actions which are not part of the testing procedure).
Kits intended for specimen collection are classified according to the kit’s intended purpose and implementing rule 1.9. Typically these kits are classified as class A as they contain a specimen receptacle (class A) and other possible IVD components likely to be class A (e.g. buffers).
Specimen receptacles and kits intended for specimen collection may be components of a kit intended for testing. They may be CE-marked and classified in their own right according to their intended purpose. The whole kit intended for testing is classified according to implementing rules 1.8 and 1.9: the intended purpose of the kit will determine its classification.
Specimen receptacles and kits intended for specimen collection may be placed on the market separately but intended by the manufacturer to be used in combination with another IVD. In this case implementing rule 1.2 applies: the specimen receptacle or kit intended for specimen collection and the other
IVD should be classified independently.
Regarding specimen receptacles and kits intended for specimen collection intended to be used by lay persons:
– Specimen receptacles or kits intended for specimen collection intended for lay persons and placed on the market separately from the testing device are not considered devices for self-testing.
– A specimen receptacle or a kit intended for specimen collection may be a component of a kit intended for testing in which the lay person not only collects the specimen but also performs one or more actions that are part of the testing procedure. In this case, the whole kit intended for testing is considered a device for self-testing (see Rule 4). As components of the kit intended for testing, the specimen receptacle or specimen collection kit may be CE-marked and classified in their own right according to their intended purpose and would not themselves be considered devices for self-testing.
As a reminder, according to Article 1(3)(b), invasive sampling products or products which are directly applied to the human body for the purpose of obtaining a specimen are themselves not IVDs.
EXAMPLES (non-exhaustive)
– A standalone urine collection cup, a stool container, a saliva collection tube or a blood spot collection card (e.g. specimen collection via finger-prick) intended for subsequent in vitro diagnostic examination are in class A.
– Standalone kits intended for the collection of saliva by the lay person for the purpose of detection of SARS-CoV-2 (by another device placed on the market separately) are in class A.
– Standalone kits intended for the collection of stool by the lay person for the purpose of faecal occult blood detection in colorectal cancer screening by a professional laboratory (with another device placed on the market separately), including a paper sheet to collect stool, a plastic stick to collect samples and a pre-filled tube for conservation and transport are in class A.
RULE 6
Devices not covered by the above-mentioned classification rules are classified as class B
RATIONALE
Rule 6 applies to devices not covered by rules 1-5.
An erroneous result with these devices is unlikely to have a significant negative impact on patient outcome, cause death or severe disability or put the individual in immediate danger.
Depending on the intended purpose, this rule would likely capture clinical chemistry tests for hormones, vitamins, enzymes, metabolic markers, electrolytes and substrates as well as the majority of anatomic pathology immunohistochemical assays. It also includes IVDs that detect infectious agents that present a moderate risk to the individual. In case of changes in the epidemiological context, for example, a new strain of infectious agent listed below, classification rules 1 or 3 could become applicable and the class of the device revised.
EXAMPLES (non-exhaustive)
– Device intended to detect and measure magnesium to assess electrolyte / magnesium homeostasis.
– Test intended to detect and measure C-reactive protein or calprotectin to detect systemic inflammatory processes due to an active disease.
– Biochemical test for establishing the identification of microbiological culture isolates or for determining antimicrobial susceptibility of microbiological culture isolates except those permitting identification or determination of MIC associated with a life threatening condition.
– Test to detect Helicobacter pylori, Clostridium difficile, adenovirus, rotavirus and Giardia lamblia.
– Non-typhoidal anti-salmonella antibodies to detect the exposure to an infectious agent.
– FSH device for fertility testing in blood.
– Device intended for the detection of Candida albicans.
– Device intended for the detection of or exposure to Entamoeba histolytica.
– Device intended for the detection of Sarcoptes scabiei (genital scabies).
– Assay intended for the detection of autoantibodies (e.g. anti-sm/RNP and anti-SSA/Ro) associated with systemic lupus erythematosus (SLE), anti-neutrophil cytoplasmic antibodies [ANCAs] in systemic vasculitis), anti-aquaporin-4 antibodies (anti-AQP4) in neuromyelitis optica spectrum disorders (NMOSDs) or organ-specific autoimmune diseases (e.g. anti-Insulin antibodies in insulin-dependent diabetes).
– Antibody tests for HAV, dengue, chikungunya and West Nile virus.
– Assay intended for the detection of IgG antibodies against HEV.
– Device intended for the detection of Influenza A/B virus (not highly virulent).
– Device intended for the detection of SARS-CoV-2
– Device intended for the detection of antibodies against SARS-CoV-2
RULE 7
Devices which are controls without a quantitative or qualitative assigned value are classified as class B
RATIONALE
Rule 7 applies to controls which are described as un-assayed where control values are assigned by the user and not the manufacturer. The manufacturer may indicate whether a specific analyte is present or absent in these controls without indicating expected assay results.
Controls with quantitative or qualitative assigned values are classified according to implementing rule 1.6.
As a reminder, and according to Article 1 paragraph 3(c) and 3(d), “internationally certified reference material” and “materials used for external quality assessment schemes” are not IVDs.
EXAMPLES (non-exhaustive):
– Unassigned control sera.
– Control materials used to verify the migration of immunochromatographic assays.
– Unassigned QC Material as a heterozygous quality control to monitor analytical performance of the extraction, amplification and detection.
– Non-assay specific control plasmas for use in coagulation.
– Non-assay specific control serum containing multiple biochemical analytes.
– A DNA or RNA probe supplied for use as a non-assay specific normal control for in situ hybridisation (ISH).
Annex I: Examples of classifying IVDs used in combination
Example 1: Enzyme-linked immunosorbent assay (ELISA) and analyser; where the analyser and its software are intended to run the assay and measure the output.
List of devices used in combination: Analyser and the software driving and influencing it, reagents, calibrators, controls, buffer/ washing solutions.
NOTE: This example is intended to illustrate how classification depends on the intended purpose of the device and highlights where the implementing rules apply to devices used in combination.
- Analyser and the software driving and influencing it which can be made available separately: In this example, the analyser is intended to only run and measure the output from an ELISA assay. The analyser and the software driving and influencing it are classified as Class A under rule 5b and implementing rule 1.4.
- Reagents: Used with the above analyser, reagents intended to assay one or more markers are classified in their own right. These reagents are classified as Class B, C or D depending on the intended purpose of the reagent.
- Calibrators: calibrators intended to calibrate the ELISA assay reagents. The calibrators are classified in the same class as the reagents for which they are acting as calibrators e.g. Class B, C or D.
- Controls:
- Controls with quantitative or qualitative assigned values intended to act as controls for a particular set of reagents are classified in the same class as the reagents per implementing rule 1.6. e.g. Class B, C or D.
- Controls without quantitative or qualitative assigned values are classified as Class B per rule 7.
- Buffer/ washing solutions: Where the buffer/washing solution possesses no critical characteristic it is classified as Class A per rule 5a.
Example 2: Blood Gas Analyser and associated devices.
List of devices used in combination: Blood gas analyser and the software driving and influencing it, sensor cassette/reagents, calibrators, controls, software, solution pack/buffers.
NOTE: This example is intended for illustrative purposes only and does not provide an exhaustive classification for all Blood Gas Analysers and associated devices. Different classes may apply depending on the intended purpose of the device.
- Blood gas analyser and the software driving and influencing it are classified as either
- Class C (Rule 3j) where the blood gas analyser directly measures a parameter from a specimen (in the absence of a reagent or disposable sensor cassette) AND where an erroneous result in a measured parameter WILL LEAD to a patient management decision resulting in a life-threatening situation.
- Class B (Rule 6) where the blood gas analyser directly measures a parameter from a specimen (in the absence of a reagent or disposable sensor cassette) AND where an erroneous result in a measured parameter WILL NOT LEAD TO a patient management decision resulting in a life-threatening situation.
- Class A (Rule 5b) where the analyser does not directly measure a parameter from a specimen, rather the specimen is analysed using replaceable sensor cassette / reagents.
- Sensor cassette / reagents are classified as either
- Class C (Rule 3j) where the blood gas analyser examines a specimen using replaceable sensor cassette / reagents AND where these sensor cassette /reagents measure at least one parameter where an erroneous result will lead to a patient management decision resulting in a life threatening situation.
- Class B (Rule 6) where the blood gas analyser examines a specimen using replaceable sensor cassette / reagents AND where these sensor cassette/reagents DO NOT measure any parameter where an erroneous result will lead to a patient management decision resulting in a life-threatening situation.
- Solution pack is classified as either
- Class C (Rule 3j) where the solution pack possesses critical characteristics and contains reagents which are integral to the measurement of an analyte AND where an erroneous result for this analyte WILL LEAD to a patient management decision resulting in a life threatening situation.
- Class B (Rule 6) where the solution pack possesses critical characteristics and contains reagents which are integral to the measurement of an analyte AND where an erroneous result for this analyte WILL NOT LEAD to a patient management decision resulting in a life threatening situation.
- Class A (Rule 5a) where the solution pack is used in conjunction with the analyser and the solution pack does not possess any critical characteristics and does not contain any reagents which are integral to the measurement of an analyte.
- Calibrators are classified in the same class as the device. Calibrators which are used to calibrate the response of the sensor cassette/ reagents are classified as
- Class B or C depending on the classification of the device (sensor cassette/reagent) being calibrated.
NOTE: Where calibrators are used to calibrate the response of an analyser and where the analyser directly measures the specimen without the use of any additional disposable sensor cassette/reagent then the calibrator is the same class as the analyser.
- Controls are classified in the same class as the device they are acting as control for e.g. the sensor cassette/reagent or the analyser.
- Where the control does not have any quantitative or qualitative assigned value it is classified as Class B under rule 7.
NOTE: Where controls are used for the purposes of assessing the response of an analyser and where the analyser directly measures the specimen without the use of any additional disposable sensor cassette/reagents then the control is in the same class as the analyser.
Annex II: Flowchart to help determine whether an IVD is a CDx
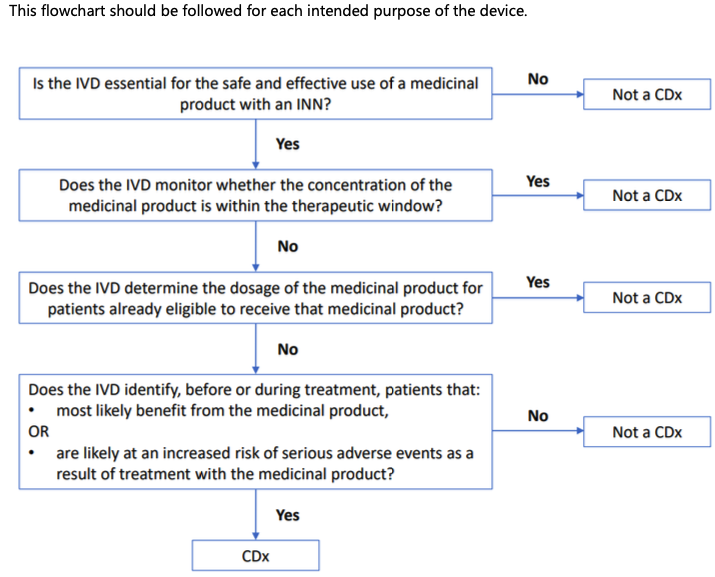
Footnotes
(1): Modified from Guidance for the Risk-based Classification System for In vitro Diagnostic Devices (IVDDs), Health Canada, 2016
(2): Article 2(5) of Regulation (EU) 2017/746 introduces significant changes to the definition of a device for self testing, please familiarise yourself with these important changes.
(3): DIRECTIVE 2002/98/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 27 January 2003 setting standards of quality and safety for the collection, testing, processing, storage and distribution of human blood and blood components and amending Directive 2001/83/EC
(4): COMMISSION DIRECTIVE 2006/17/EC of 8 February 2006 implementing Directive 2004/23/EC of the European Parliament and of the Council as regards certain technical requirements for the donation, procurement and testing of human tissues and cells
(5): DIRECTIVE 2010/45/EU DIRECTIVE 2010/45/EU (corrigendum: 2010/53/EU) OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 7 July 2010 on standards of quality and safety of human organs intended for transplantation
(6): The IVDR requires the INN to be provided in the instructions for use of the CDx. In case several INNs are linked to a single CDx, all INNs should be listed in the IFU.
(7): On the topic of biochemical genetic tests, according to the wording of rule 3i, it was determined that these tests may be classified by this indent but may be better classified under other rule indents depending on the intended purpose. Biochemical genetic tests are considered to be tests which provide information on the amount or activity level of proteins; where abnormalities in these proteins result from a genetic disorder, for example, for inborn errors of metabolism.
(8): Useful links to some national and EU screening programmes:
– http://www.screening-dgns.de/reports.php#weitere
– https://ec.europa.eu/search/?query_source=PUBLICHEALTH&QueryText=new+born+screening&op=&swlang=en&form_build_id=form-i3HYd2vGVLMjC0BcN2Zvog1lGZ7jf5DgYdXQ5vWr70w&form_id=nexteuropa_europa_search_search_form
(9): within the meaning of Directive (EU) 2015/1535 of the European Parliament and of the Council, OJ L 241, 17.9.2015, p. 1.
Revision History
Redline Version
Redline Version
Redline Version
Redline Version