
MDCG 2019-8 Rev.2
Implant Card relating to the application of Article 18 Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices
Disclaimer: This document is an interactive version of the original MDCG document. We will keep it up-to-date.
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.
Table of Contents
1. Scope
This document provides guidance for Member States, concerned industry and other stakeholders on a blueprint of an implant card (IC) required by the MDR (Regulation (EU) 2017/745). It describes the intended use, content and information to be provided by the manufacturer together on the IC and a definition of fields to be completed by the implanting healthcare institutions or healthcare providers according to national law in Member States. Whereas the intended purpose and most of the data elements of the IC are already defined in Article 18 of the MDR, this document contains the description of other data elements which must be completed by the healthcare institution or healthcare provider and which must be considered by the individual Member State when implementing Article 18 MDR as required (1).
2. Purposes of the Implant Card
The aim of introducing an IC has been to achieve three main objectives:
- Enable the patient to identify the implanted devices and to get access to other information related to the implanted device (e.g. via EUDAMED, and other websites).
- Enable patients to identify themselves as persons requiring special care in relevant situations e.g. security checks.
- Enabling e.g. emergency clinical staff or first responder to be informed about special care/needs for relevant patients in case of emergency situations.
3. Legal consideration on Implant Card design
Article 18 MDR describes the requirements regarding the IC which shall be provided by the manufacturer together with the device. Whereas Article 18 para 1a) describes the information which shall be provided by the manufacturer on the IC, Article 18 para 2 lays down the obligation of Member States to require health institutions to provide the IC to the concerned patient.
In accordance with Article 18, 1a) the manufacturer should provide the following information on the IC (preferably on the card itself or, alternatively, as stickers to be placed by the clinician):
- Device name;
- Serial number, lot number;
- Unique device identification (UDI);
- Name, address and the website of manufacturer;
- Device type. (2)
Article 18, 2 lays down that Member States shall require health institutions (or healthcare providers) inter alia to make available the IC to the relevant patient. The IC should bear their identity. For this purpose, the IC provided together with the device should contain blank fields which shall be filled out by the health institution or healthcare provider, respectively.
Since the establishment of (and the logistics behind) many different national IC designs is very expensive and of no additional benefit, Member States should ensure that, in the context of national implementation of Article 18 of the MDR, only the following information is required to be provided by the health institution or healthcare provider on the IC (together with the information provided by the manufacturer). Accordingly, a European or international blueprint for an IC which supports the purposes described in Article 18, 2 should contain the following blank fields:
- Name of the patient or patient ID;
- Name and address of the health institution or healthcare provider who performed the implantation;
- Date of implantation.
In order to be fit for the purposes described in Article 18, the outer dimensions of the IC should be the same as those of credit cards, ATM cards or ID cards (85.60 mm × 53.98 mm /3 3⁄8 × 2 1⁄8 inches) and with a radius of 2.88–3.48 mm (3).
4. Information to be provided by the manufacturer on the Implant Card and information to be added by the health institution
The manufacturer shall provide the following necessary information:
- Device name;
- Device type;
- Serial number or, where applicable, lot or batch number;
- Unique device identification (UDI); the UDI as AIDC (4) format and the UDI-DI as HRI (5)
- Name and address of the manufacturer of the medical device;
- Website of the manufacturer of the medical device.
The text provided on the IC and on the instruction for completing the IC by the healthcare institution or healthcare provider must be legible and at least 2 millimetres high. ‘Text’ includes any: number, letter, symbol, or letter or number in a symbol.
In addition, the manufacturer should design the IC in a way that the following blank fields to be filled out by the implanting healthcare institution or healthcare provider are available:
- Name of the patient or patient ID;
- Name and address of the healthcare institution which performed the implantation;
- Date of implantation.
5. Use of symbols
To avoid national versions of the IC, the use of symbols is advisable.
The following list contains symbols which have either been validated by users and have been submitted and accepted for inclusion in an upcoming international standard or already exist in the ISO database (Online Browsing Platform). The explanation of symbols on the IC should be provided in a leaflet (see section 7) or on the back of the IC, if space allows.
Article 18(1) lays down that the information provided in the IC shall be stated in the language(s) determined by the concerned Member State.
List of symbols recommended for use on the IC (6):
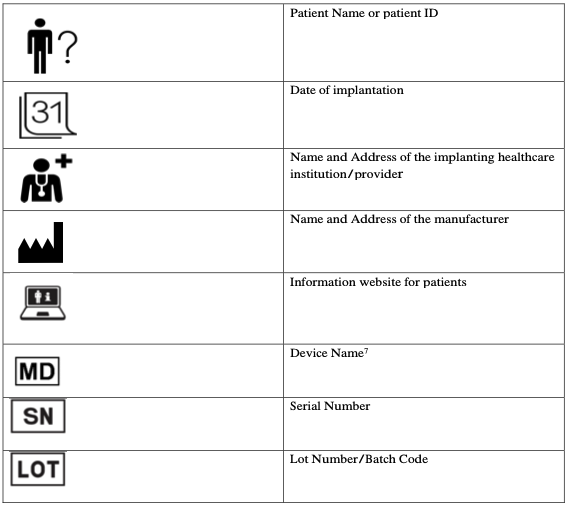
UDI-DI information in a HRI format should be introduced by the wording “UDI-DI”.
6. Language requirements on specific fields
Despite the nearly complete list of symbols for fields on the IC, there is currently no symbol available for the required field “Device Type”.
The lack of a symbol and the purpose of this field makes it necessary that the information on the device type must be provided in the language accepted/required by the concerned Member State.
There are several possibilities available to provide this information in the necessary languages, e.g. the information is already printed on the IC in the different languages or stickers are provided with the IC and the healthcare professional selects the right one etc.
7. Benefits of an informative instruction leaflet
(8)
As mentioned above (section 5), there is a need to provide, together with the IC, instructions on how to complete the IC and to explain the used symbols. This information shall be stated in the language(s) determined by the concerned Member State. For this reason, the use of a leaflet, containing the relevant information, to be provided together with the IC and the implantable device, is the recommended solution.
As part of the risk management, the manufacturer has to investigate, by means of an ergonomic analysis or ergonomic usability test procedure, if the provided instructions are sufficient to enable the health professional to complete the IC correctly. The instructions must reflect the solution chosen by the manufacturer (e.g. pre-printed IC, blank IC with sticker(s) to be added, mixture of both). Special considerations might be needed when providing a system IC or an IC for a separate implantable component, when there is not yet a common practice (standard) established.
8. Implant Card for implantable systems
If an implantable device contains implantable components which might be replaced by other (or the same) components, for example in case of a later revision, the manufacturers should consider the use of a System IC. In Annex I to this guide an example is provided.
Ways could be explored by relevant stakeholders to develop common rules on how the necessary information to be placed on the System IC is delivered with the replaceable component and how health professionals could ensure that the System IC is appropriately updated, when necessary.
Annex I examples of principle designs of Implant Cards and leaflets
GENERAL NOTE: All examples contained in this Annex are to be intended as illustrative examples only. (9)
The basic shape of the IC shall be designed in compliance with ISO/IEC 7810 ID-1 (credit card form).
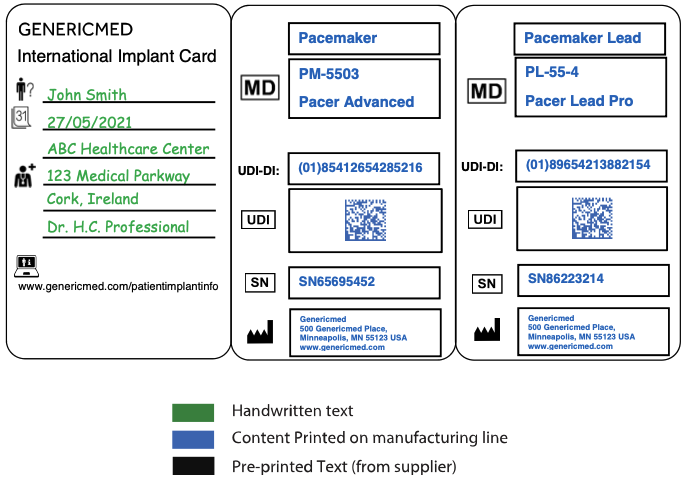
The following pictures provide examples for individual device ICs.
Example 1:


Example 2:

Example 3 (suggested design of a foldable System IC):
To be able to represent medical device systems in one IC, the IC shall be available in collapsible form.
Examples of IC Leaflet
Front 1 (no stickers – preferred option)

Front 2 (sticker only for device type information)

Front 3 (stickers)

Back
Footnotes
(1): This guidance doesn’t include the patient information described in Article 18 para 1 (b-d) which might be provided by the manufacturer by any means that allow rapid access to that information and that shall be stated in the language(s) determined by the concerned Member State.
(2): In Article 18 1a) of the MDR, the term “device model” is used. However, this term which is not defined in the MDR, is part of the UDI-DI related information to be provided to EUDAMED. To avoid duplication and in the context of the intended purpose of the IC (to be used in situations requiring special care or in emergency situations), it is considered that reference to device model in the MDR for the purpose of the IC shall be read as reference to device type, like “pacemaker”, “hip implant”, etc. When the European Medical device nomenclature is made available, a “standardised” set of terms of this nomenclature should be recommended for use in relation to the field “device type”.
(3): Dimensions to conform to the standard ISO/IEC 7810 ID-1.
(4): AIDC – Automatic identification and data capture format (e.g. linear or 2D-Barcodes)
(5): HRI – Human readable interpretation
(6): The symbols for patient name or patient ID, name and address of the implanting healthcare institution/provider, date of implantation, name and address of the manufacturer, serial number, lot number/batch code are already available and published in existing ISO standards. The symbols for device name, patient information website and UDI have been validated by users according to the ISO 15223-2 process. Note that the symbols listed here to indicate the following terms: ‘Device Name’, ‘Patient Name or Patient ID’, ‘Date of Implantation’, ‘Name and address of the implanting healthcare institution/provider’ on the implant card, are used in the ISO context to indicate ‘Medical Device’, ‘Patient Identification’, ‘Date’, ‘Health Care Center or Doctor’.
(7): Please note that in the ISO context, the ‘MD’ symbol is used to identify that the product in question is a medical device. On the implant card, this symbol is used to indicate the device name.
(8): This leaflet should not be confused with any possible vector of information referred to in points “b”, “c” and “d” of Article 18(1) of the MDR.
(9): Please note that this includes translations (into languages other than English) shown in Examples throughout Annex 1, which are machine translated and not intended to be read as official translations into EU languages.
Revision History
March 2020
Redline Version
Redline Version